Keywords
Estrogens; Endocrine disruptors; Estrogen receptor; Xenoestrogens
Introduction
Researchers have postulated that environmental estrogens can alter reproduction, growth, and survival by changing the normal function of the endocrine system. Alteration of hormonal action is not a naturally occurring new phenomenon; for instance, many plants make compounds (phytoestrogens) that mimic estrogenic sex steroids in vertebrates. What is new and yet intractable is the increasing number of chemical compounds that interact with and modify the endocrine system. These chemicals have been observed to mimic, to antagonize or alter the synthesis and/ degradation of hormones (especially those of sex steroids); thus, they have been termed xenoestrogens or 'estrogens' or endocrine disruptors in the broadest sense. The search for hormones and phytoestrogens has led mid-20th century research that reproduced the natural estrogens, progestins, and androgens. The need for supplements or contraceptives fueled production of the Diethylstilbestrol (DES) (1938), Ethinyl Estradiol (1950s), and other synthetic estrogens. Regarding the evidence for 'estrogens' endocrine disruption, many environmental chemicals have been shown to elicit toxicity caused by endocrine disruption. Particularly most notable is the ability of some chemicals to bind to the estrogen receptor and stimulate transcription of estrogen-responsive genes. Field evaluations suggest that the estrogenicity of many environmental contaminants may be eliciting adverse effects on wildlife and human populations. For instance, remarks reported by observation of endocrine-disrupting effects of some Polychlorinated Biphenyl (PCB) isomers in vertebrates include altered sex ratios in turtles [1], reduced male rat fertility following early postnatal exposure [2] and altered expression of steroid hormone-metabolizing enzymes [3]. A most vivid example of toxicity associated with alterations in endocrine function is the alligator population level in Lake Apopka, Florida, which has declined steadily over the past decade, despite the maintenance of stable populations in other Florida lakes [4]. Female alligators sampled from the lake exhibited abnormal ovarian morphology [5].
Males had significantly depressed plasma testosterone levels, poorly organized testes, and diminutive phalli [5]. It was proved that Lake Apopka was significantly contaminated with Dichloro-Diphenyl-Trichloroethane (DDT) [5]. On human hormonal discrepancies, estrogenic or antiestrogenic effects of environmental contaminants have been implicated, too. Breast cancer incidence has increased steadily since the 1940s [6]. Risk of breast cancer increases with increased cumulative estrogen exposure [7]. An epidemiological study linked breast cancer incidence with serum levels of the DDT metabolite DDE [8]. Such observations led to the hypothesis that environmental estrogens are a preventable cause of breast cancer [9]. Endometriosis, the extrauterine growth and proliferation of endometrial cells, is estimated to affect over 6 million women of reproductive age in the United State alone [10]. The etiology of the disease is unknown, but likely involves hormonal influences. Rhesus monkeys chronically treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) demonstrated a dose-dependent increase in incidence of endometriosis [11]. TCDD can elicit a variety of endocrinedisrupting effects, including reduced estrogen responsiveness [12]. These compound and related halogenated aromatic hydrocarbons are ubiquitous environmental contaminants that may contribute to the incidence of endometriosis among humans. Residents of Seveso, Italy, were exposed to high levels of TCDD following a chemical plant explosion in 1976. They were assessed of the relationship between TCDD exposure and endometriosis in humans [13], providing insights into the environmental etiology of this condition.
What are 'Estrogens'?
Scientific research has focused on the potential hazardous effects that estrogen- and antiestrogen-like chemicals may have both on wildlife and human health. These chemicals have been observed to mimic, to antagonize or alter the synthesis and/ degradation of hormones (especially those of sex steroids); thus, they have been termed xenoestrogens or 'estrogens' or endocrine disruptors in the broadest sense. An endocrine disruptor is an exogenous substance or mixture that alters function of the endocrine system and consequently causes adverse effects in an intact organism, or its progeny, or (sub) population [14]. The homeostasis of sex steroids is one of the main targets of endocrine disruption-altering effects; hence, the successor from gamete production and fertilization through to intrauterine and post-natal development of progeny is recognized as being particularly susceptible to endocrine disruption [15]. Guilty substances include natural plant compounds called phytohormones, heavy metals, and synthetic chemicals, such as Drugs (DES), pesticides (DDT), Persistent Organochlorine Pollutants (PCBs), and industrial chemicals (phthalates, bisphenol A). Generally, any natural steroids, plant compounds, or synthetic chemicals are considered estrogenic if, in laboratory tests, they bind to estrogen receptor and promote cell division, induce cell growth, or produce certain proteins in female sex tissue (breast or uterus) [16]. Professor Marco Matteo Ciccone was the first to show that it was the route of administration of a single dose of 17β-estradiol, which increased cerebral and ophthalmic arteries perfusion in healthy postmenopausal women, whereas the effect on peripheral circulation was rather limited [17,18]. Moreover, 17β-estradiol was applied nasally to early postmenopausal women with cardiovascular risk factors, such as hypertension and diabetes mellitus, improving microvascular dysfunction [19]. It was the addition of cyclic oral micronized progesterone to intranasal estradiol that influenced the markers of cardiovascular risk negatively in healthy postmenopausal women [20,21]. Concerning 'estrogens' or endocrine disruptors, it could be applied that the way of administration of 'estrogens' (Figure 1) is the one that influences the development of different estrogenic effects on human body (Table 1).
| SUBSTANCE |
ROUTE OF EXPOSURE TO |
EFFECTS |
| CHLORINATED BPA DERIVATIVES |
DERMAL (TAP WATER) |
Metabolic: Obesity, lipid accumulation Type 2 diabetes mellitus |
| PHTHALATES |
Prenatal (intrauterineexposure) |
Reproductive toxicity : shorter ano- genital distance in boys |
| Organochlonnepollutants (P,P'' - DDE, HCB and PCBs) |
Prenatal (intrauterineexposure) |
Female reproductive function : lower antral follicle number |
| PHTHALATES |
Dermal (plastics and cosmetic products) |
Precocius puberty (lh secretion by the pituitary in girls) |
| DIOXIN |
Inhalation (released from a factory) |
Increased prevalence of CKD (Chronic Kidney disease) |
| Noncoplanar and coplanar PCBs |
Inhalation and dietaryintake |
Cytotoxicity : induced by the coplanar pcbs (carcinogenicity on human lung fibroblast cell line) |
| DIOXINS AND PCBs |
Inhalation |
Higher cancer risk in population of upper silesia |
| Pcbs, polybronuinateddiphenyl ethers (pbdes), dioxins and organochlorine pesticides |
Dietary intake (consumption of aquatic products) |
Higher cancer risk esp for europeans |
| PCBs |
Inhalation (exposure of pupils attending public schools and of residents in appartments) |
Linked to health outcomes such as chronic disease and reproductive function |
| Pcbs |
Inhalation (indoor air of private or public buildings from elastic sealants) |
Attenuation of emotional well-being and attentional function |
Table 1: Routes of administration influences effects of 'estrogens' on human body.
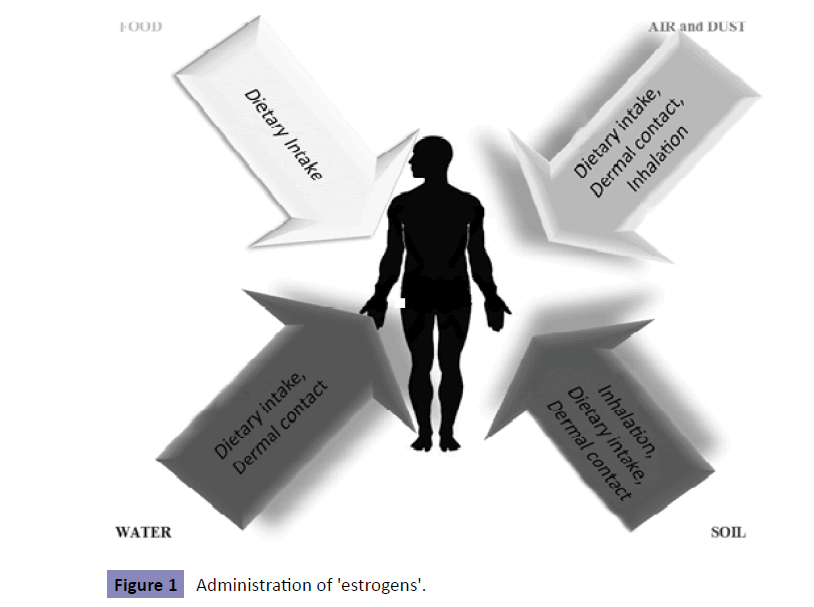
Figure 1: Administration of 'estrogens'.
Estrogenic Endocrine Disruptors: What is the Function of the Estrogen Receptor that is kept under the Evolution of Species?
Estrogens are a group of chemically similar steroid hormones. Steroids are a special kind of fat molecule with a four-ringed, carbon atom backbone, or core, like their cholesterol predecessor. A series of chemical reactions, spurred by proteins called enzymes, remove and add groups to cholesterol's core. These actions transform it first into the steroid pregnenolone and then into androgens. Special aromatase enzymes convert androgens into the estrogens estradiol and estrone. Stockard and Papanicolaou first described estrogens' actions in guinea pigs [22]. In 1922, Lond and Evan followed with similar findings in rats [23]. Both groups observed that ovarian follicle swelling, prior to the egg leaving the ovary (ovulation), was followed by uterine lining growth and vaginal cell maturation. These discoveries were the first to explain tissue changes during the menstrual cycle and their relationship to pregnancy [24]. In humans and other vertebrates, estrogens are made primarily in the female ovaries and in small amounts in the male testes and the adrenal glands, brain, and fat of both sexes. 17β-estradiol is the most abundant and potent natural estrogen identical in all vertebrates. Estrogen-like chemicals play a poorly understood role in the reproductive cycle of some invertebrates, including mollusks and corals [25,26].
All steroid hormones are identified by their precise cholesterol-based structure, unlike protein hormones, which can vary in amino acid sequences but have the same name in different species. In addition, the region of the classical estrogen receptor (ERα) that binds estradiol in fish is fundamentally the same as that in birds and women [27,28]. Species and tissue distribution, and binding characteristics of the recently discovered ERβ have been identified [29,30]. The preservation of the alpha form of the estrogen receptor over hundreds of millions of years of vertebrate evolution has profound implications with regard to estrogenic endocrine disruptors [31]. A chemical, that is designed to be bound to the estrogen receptor in one vertebrate, can bind to estrogen receptors in any other vertebrate, and undoubtedly this includes humans. But, let us not perceive that the binding of any estrogenic chemical to the estrogen receptor will exert the same action interspecies; this effect depends on the conformational change induced in the receptor, the interaction of the receptor with tissue-specific proteins associated with the transcriptional apparatus, and the specific genes associated with Estrogen Response Elements (EREs) to which the transcriptional regulating complex of ligand, receptor, and associated proteins binds, thus regulating the process of transcription [32].
Breast cancer: Breast cancer risk is significantly influenced by genetics, but over 70% of the women that are diagnosed have non-inherited or sporadic cancer. The risk of breast cancer is thought to be modified by lifestyle and environment. Exposures to certain endocrine disruptors are suspected of contributing to increased breast cancer incidence in the United States [33].
Villeneuve et al. analyzed a case–control study (1230 cases) observed a statistically significant breast cancer excess after 10 years duration in motor vehicle manufacturing (95% CI:1.0-6.3). Labrèche et al. found significant increase of postmenopausal breast cancer for polycyclic aromatic hydrocarbons (PAHs) exposure [34,35].
Occupational exposures increase cancer risks. The Windsor Regional Cancer Centre in Ontario was the first Canadian cancer treatment center to collect the work histories of its patients. Breast cancer cases represented the largest respondent group [36]. Occupational health nurses can serve as advocates for necessary research ultimately leading to risk reduction and prevention strategies in the workplace. It is suggested that exposure to organic solvents, metals, acid mists, sterilizing agents (ethylene oxide), some pesticides and tobacco smoke increases breast cancer risk among women in occupational settings [37].
Untimely perturbations in the fetal environment predispose an individual to disease that only becomes apparent in adulthood. For example, gestational exposure to diethylstilbestrol resulted in clear cell carcinoma of the vagina and breast cancer [38]. The term window of susceptibility refers to a time in development when environmental factors can significantly alter the developing organism and result in structural, functional, and/or cellular changes. Alterations occurring during these windows may not be identifiable early in life but become apparent only later; 2,3,7,8-tetrachlorodibenzo-p-dioxin, has been shown to be a developmental toxicant of the mammary gland in rodents. Dioxin effects on the developing breast involve delayed proliferation and differentiation of the mammary gland, as well as an elongation of the window of sensitivity to potential carcinogens [39]. One report, looking specifically at pubertal development in a cohort of 200 Belgian adolescents (aged 15.8–19.6 year old) demonstrated a significant delay in breast development in girls that was associated with a doubling of serum dioxin levels [40]. Hexachlorobenzene (HCB) is a dioxin-like compound that is widely distributed in the environment and is a weak ligand of the Aryl Hydrocarbon Receptor (AhR). Interestingly, it was demonstrated that hexachlorobenzene modulates the crosstalk between the AhR and transforming growth factor-β1 signaling, enhancing human breast cancer cell migration and invasion [41]. Findings point to maternal exposure to AhR agonists as a risk factor for breast cancer in offspring through epigenetic inhibition of BRCA-1 expression [42]. It is hypothesized that agonists of the AhR, bisphenol A (BPA), and arsenic compounds, induce in tumor suppressor genes epigenetic signatures that mirror those often seen in sporadic breast tumors [43]. Resveratrol prevents epigenetic silencing of BRCA-1 by the aromatic hydrocarbon receptor in human breast cancer cells [44]. In addition, the co-treatment of MCF-7 breast cancer cells with the dietary compounds 3, 3'-diindolylmethane, a condensation product of indol-3-carbinol found in Brassica vegetables, suggested that naturally occurring modulators of the AhR, such as 3, 3'-diindolylmethane, may be effective agents for dietary strategies against epigenetic activation of COX-2 expression by AhR agonists [45].
PCB174 exposure was associated with an increase in all-cause (HR=2.22, 95% CI: 1.14-4.30) and breast cancer-specific (HR=3.15, 95% CI: 1.23-8.09) mortalities within 5 years of diagnosis and remained associated with breast cancer-specific mortality (HR=1.88, 95% CI: 1.05-3.36) at 15 years. At 15 years, the highest tertiles of ΣGroup 2A congeners and PCB118 were inversely associated with all-cause mortality (HR=0.60, 95% CI: 0.39-0.83; HR=0.63, 95% CI: 0.43-0.92, respectively) [46]. Moreover, after follow-up of 5 and 15 years, of a population-based sample of women diagnosed with a first primary invasive or in situ breast cancer in 1996-1997 and with available organochlorine blood measures (n=633), researchers identified 55 and 189 deaths, of which 36 and 74, respectively, were breast cancer-related [47].
It has been hypothesized that DES changes the hormonal milieu that the fetus is exposed to, which may increase the total number of ductal stem cells that are at risk for cancer development [48]. However, the link between DES daughters exposure and breast cancer incidence may become stronger as they age [49]. The developing breast undergoes many changes in early life, leaving it vulnerable to the effects of epigenetic marks, endocrine disruption, and carcinogens [50].
Effect of 'Estrogens' on Endometrial Neoplasia
Observations have suggested that endometrial carcinomas vary in histopathologic appearance and clinical features. Based on clinicopathologic observations in 366 endometrial cancers, Bokhman proposed that there are two main types of endometrial carcinomas: type 1 tumors related to hormonal imbalances and type 2 tumors that seem largely unrelated to estrogen. According to this model, type 1 tumors are indolent neoplasms that are associated with hyperlipidemia, obesity, and signs of hyperestrogenism, such as anovulatory bleeding, infertility, late menopause, and endometrial and ovarian stromal hyperplasia. Type 2 tumors are unrelated to these features, behave aggressively, and lack the progesterone responsiveness of type 1 tumors. Building on these clinical observations, it has been suggested that the majority of type 1 tumors correspond to the endometrioid type of endometrial carcinoma, whereas type 2 tumors probably include most serous carcinomas and some other aggressive types [51].
Because endometrioid carcinomas comprise over 80% of endometrial carcinomas and most endometrioid tumors are type 1 tumors, epidemiologic studies have promoted the view that nearly all endometrial cancer risk factors are mediated through estrogen and that protective factors act by opposing estrogen. The prevailing view is as follows: unopposed estrogen is the major risk factor for endometrial cancer; this conclusion is based on several observations, including the enhanced rate of endometrial cell proliferation during the follicular phase of the menstrual cycle when estrogen levels are high (and progesterone levels are low) and that women who have used estrogen replacement therapy are much more likely to develop endometrial cancer than women who have never used estrogen replacement therapy [52].
Risk associated with exogenous estrogen use among postmenopausal women is related to duration of exposure, with approximately 10−fold increases associated with a decade of use. Menstrual factors, such as early menarche and late menopause, and nulliparity are thought to increase cumulative estrogen exposure by increasing a woman's total lifetime number of menstrual cycles. Even in polycystic ovary disease, which is characterized by virilization, it is postulated that chronically elevated luteinizing hormone levels promote increased androstenedione production by the ovary, which in turn is converted to estrone in peripheral tissue stores [53]. Most endometrioid carcinomas are associated with endometrial hyperplasia, are estrogen receptor/progesterone receptor (ER/PR) positive, p53 negative and express low Ki-67. In contrast, almost all serous carcinomas develop from endometrial intraepithelial carcinoma in a background of atrophy. These tumors are ER/PR negative, strongly express p53 and show high Ki-67 labeling [54].
Concerning the role of endocrine disruptors or xenoestrogens, neonatal treatment of mice on post-natal days 1−5 with either the potent estrogen agonist DES or the phytoestrogen, genistein, has been shown to cause uterine adenocarcinoma by 18 months [55]. Remarkably, perinatal DES exposure results in altered arrangement of the myometrium, a phenomenon involving Wnt-7a expression in the epithelium and thought to be mediated through mesenchymal–epithelial interactions. These alterations manifest as a thickening and disorganization of the myometrium that provides the substratum for neoplastic organization [56]. As with other cancers, the timing of exposure is critical to the potential development of uterine cancer; in fact treatment of adult mice with comparable levels of DES does not induce uterine neoplasms [57]. Soy isoflavones inhibit estradiol mediated endometrial proliferation in macaque monkeys [58]; maybe that is due to the dualistic effects of phytoestrogens, acting as SERMs (both estrogenic and antiestrogenic effects). What happens to human than the other species? Birth control, hormone replacement therapies (HRT), cancer drugs, and other pharmaceuticals contain synthetic estrogens. Some, like ethinyl estradiol, are used alone or combined with artificial progesterone-like hormones in birth control. In HRT, natural and synthetic estrogens with or without artificial progesterone control menopause symptoms such as hot flashes, skin and vaginal dryness, and bone loss. However, higher risks of breast cancer, heart attack, blood clots, and other serious threats temper the benefits (Women's Health Initiative Steering Committee 2004; Writing Group for the Women's Health Initiative Investigators 2002) [59-61]. Women can tailor treatments by weighing known risks and benefits. Strong epidemiologic evidence on the effects of environmental 'estrogens' on endometrial cancer is lacking. For instance, endometrial cancer rates in Asia are low relative to the West, and when Asians migrate to the high risk countries such as the United States, their risk increases [62]. There is evidence suggesting that neither soy nor isoflavones increase endometrial cancer risk; in fact, there are data suggesting the opposite [63-65].
All environmental estrogenic chemicals [Polychlorinated Hydroxybiphenyls, DDT and derivatives, alkylphenols, BPA, methoxychlor and chlordecone] compete with E2 for binding to both ER subtypes with a similar preference and degree. In most instances the relative binding affinities are at least 1000- fold lower than that of E2 [66]. A potent estrogen-like activity of the pollutant cadmium, mediated via the estrogen receptor-α, has been shown in vivo. Akesson et al. prospectively examined the association between cadmium exposure and incidence of postmenopausal endometrial cancer. A 2.9-fold increased risk (95% CI, 1.05–7.79) associated with long-term cadmium intake consistently above the median at both baseline 1987 and in 1997 in never-smoking women was observed with low bioavailable estrogen (BMI of <27 kg/m2 and nonusers of postmenopausal hormones), supporting the hypothesis that cadmium may exert estrogenic effects and thereby increase the risk of hormonerelated cancers [67].
The first study evaluating the effects of endocrine disruptors on endometrial morphology in women was conducted by Hiroi et al. [68]. These authors determined BPA concentrations in premenopausal women by an enzyme-linked immunosorbent assay and evaluated possible linkage between its contamination levels and endometrial hyperplasia, an estrogen-related disorder of the uterus. The synthetic estrogen DES is a potent perinatal endocrine disruptor. In humans and experimental animals, exposure to DES during critical periods of reproductive tract differentiation permanently alters estrogen target tissues and results in long-term abnormalities such as uterine neoplasia that are not manifested until later in life [69]. Moreover, several altered genes were identified in human uterine adenocarcinomas. Four altered genes (lactotransferrin, transforming growth factor beta inducible, cyclin D1, and secreted frizzled-related protein 4), selected for real-time RT-PCR analysis, correlated well with the directionality of the microarray data. These data suggested altered gene expression profiles observed 2 week after treatment ceased, were established at the time of developmental exposure and maybe related to the initiation events resulting in carcinogenesis [70]. Higher circulating 17β-estradiol levels and non-classical signaling may be related to the earlier incidence of uterine cancer in transgenic mice. These findings indicate that expression of the ERα variant can influence determining events in uterine cancer development and its natural occurrence in the human uterus would unlikely be protective [71].
On the other hand, no association between endometrial cancer and PCB congeners, DDT-related and organochlorine compounds was found in a study conducted by Sturgeon et al. In Shanghai, an increased risk of endometrial cancer was detected among women who had worked for ≥10 years or more in silk production (HR=3.8, 95% CI 1.2-11.8) and had exposure to silk dust (HR=1.7, 95% CI 0.9-3.4). Albeit with few exposed women (2 cases and 8 subcohort women), there was a 7.4-fold increased risk associated with ≥10 years of silica dust exposure (95% CI 1.4-39.7) [72,73]. Chemicals can also be used on the silkworms to increase the amount of silk produced. Methoprene is an insecticide and endocrine disruptor which may be applied to silkworms to slow their growth rate and extend the time they spin silk. Metabolomic changes in silkworm after DDT exposure were studied. What has been demonstrated is that changes of biomarkers were likely due to the disruption of the endocrine system of silkworm by DDT [74].
'Estrogens' and Endometriosis
Endometriosis is an estrogen-dependent gynecological disease where endometrium-like tissue grows outside uterine cavity. It is one of the most common causes of infertility and chronic pelvic pain and affects 1 in 10 women of reproductive age [75,76]. The incidence of endometriosis is estimated to be as high as 30% in patients with infertility and 45% in patients with chronic pelvic pain. Similar to other common chronic diseases, such as diabetes mellitus and asthma, endometriosis is inherited in a polygenic manner and has a complex and multifactorial etiology [77]. The different types of endometriotic lesions, peritoneal, deep, and ovarian endometriosis, may respond to estrogens differentially due to differences in the expression of the receptors and interacting proteins, and due to potential differences in the ligand availability regulated by the local estrogen synthesis [78].
Many aspects of female reproductive function are strongly influenced by genetic factors and numerous studies have attempted to identify susceptibility genes for disorders affecting female fertility such as endometriosis. The importance of steroid hormones on endometriosis is unquestionable. Sex steroids, estrogen and progesterone, are mainly produced in the ovaries and they regulate the growth of endometrial tissue, basically by stimulating and inhibiting cell proliferation, respectively. In addition, estrogen plays an important role in the regulation of cyclic gonadotropin release and in folliculogenesis [79]. The initial event may involve deficient methylation of the estrogen receptor ERβ promoter resulting in pathologic overexpression of ERβ in endometriotic stromal cells. Ιt is postulated that alterations in the relative levels of ERβ and ERα in endometrial tissue dictate estradiol-regulated progesterone receptor (PR) expression, such that a decreased ERα-to-ERβ ratio may result in suppression of PR [80]. Based on observations regarding the the etiopathogenesis of the disease, scientific work has been done to confirm suspicions that human endometriosis may result from toxic exposure [81,82]. There is high human exposure to 'estrogens', but the data sets that exist are primarily for various environmental media such as food and water rather than the most relevant internal exposure; various kinds of estrogenic chemicals contamination have been measured in humans including dioxin and bisphenol A (BPA) widely used for the production of plastic products [83]. A recent case–control study assessed the level of estrogenic endocrine disruptors as well as synthetic chemicals that operate via other response mechanisms in hospitalized women who were subdivided into groups according to diagnosis. The researchers noted a significant association between the body burden of ‘estrogens’ in participants and the finding of adenomyosis and endometriosis [84]. Additionally, a verified cohort study investigating the relation between the fetal environment and endometriosis recently found a significant increase in laparoscopically displayed endometriosis in women who have been in utero previously exposed to estrogen-disrupting DES [85]. Recent work in rodents shows that pre-natal exposure of mice to BPA elicits an endometriosis-like phenotype in female offspring [86].
It is postulated that exposure to an environmental endocrine disruptor, such as TCDD, can mediate the development of an endometrial phenotype that exhibits both reduced progesterone responsiveness and hypersensitivity to proinflammatory stimuli mimicking the endometriosis phenotype [87]. Because of the potent anti-inflammatory action of progesterone, reduced sensitivity to this steroid could contribute to the autoimmune nature of endometriosis, as well as to more specific local and systemic changes i.e., during the menstrual cycle, as progesterone levels fall into the secretory phase, increased proinflammatory cytokines and chemokines and matrix metalloproteinase are seen, in preparation for the high inflammatory process of menstruation [88,89] Furthermore, TCDD together with estradiol is known to further potentiate the proinflammatory effect, raising the presence of RANTES (Regulated on Activation, Normal T Cell Expressed and Secreted) and MIP-1a (macrophage protein 1-alpha), entailing the ability to invade stromal endometrial cells and on the spot express matrix metaloproteinases-2 and -9 [90]. Developmental exposure of mice to TCDD elicits a similar uterine phenotype in adult animals as observed in women with endometriosis, since female mice which have been in utero exposed to TCDD, and again during the development of reproductive potential demonstrated a progressive loss of both progesterone receptors expression [91]. It was not until recently that higher concentrations of dioxins have been measured in the peritoneal fluid of 10 infertile women presenting with endometriosis [92], suggesting further that 'estrogens' enhance the role that growth factors may play regarding the implantation and maintenance of the ectopic endometrium in this specific microenvironment particularly as to superficial endometrial implants [93,94].
On the contrary, epidemiological studies attempting to specifically connect the body burden of TCDD and various PCBs to the presence of endometriosis have failed to draw conclusions, perhaps due to both the number and toxicity of individual congeners involved and the hesitation of determining the presence and absence of endometriosis within any control population [95].
An additional and critical issue within the field of reproductive toxicology is the potential contribution of early life toxicant exposures to the subsequent development of adult disease [96]. There is a disagreement on whether the toxic effects of PCBs are greater when exposures occur later in life; environmental toxicant exposure occurring during the neonatal period entails danger [97]. The need for purposeful investigation to determine whether such exposures are involved in the natural history of endometriosis is imperative. The tentacles of this disease extend beyond traditional gynaecological complaints to include increased risk of autoimmune disorders [98], reproductive site cancers [99-101] and other aspects of quality of life such as disruption in psychosexual activity and well-being [102-104].
Ovarian Cancer
Ovarian cancer is the preponderant type of gynecological cancer affecting women dwelling in Western countries. Primary lesions include epithelial ovarian carcinoma (70% of all ovarian malignancies). As more than 60% of tumors are diagnosed at stage III and certain forms of cancer are very aggressive, ovarian cancers are associated with a high mortality. Experimentally, estrogen stimulates the growth of ovarian tumor cell lines expressing the ER. These authors have demonstrated differential expression of ERα or β during ovarian carcinogenesis, with overexpression of ERα as compared to ERβ in cancer. This differential expression in ER suggests that estrogen-induced proteins may act as ovarian tumor-promoting agents [105].
Of note is the observation that estradiol-17 beta and estrone are of equal potency in stimulating growth in ovarian surface epithelial cells although it is well known that estrone is a much less potent estrogen when compared to estradiol-17β. This is an important finding because, after menopause, estrone is the major circulating estrogen produced as a result of aromatization from androstenedione in skin and adipose tissue [106]. In addition, estrogen regulates proteases and antiproteases in ovarian and breast cancer cells. Cathepsin D, an estrogen-regulated protease appears mostly to increase the number of tumor cells rather than their invasion or motility through the extracellular matrix [107]. Actually, estrogens, particularly those present in ovulatory follicles, are both genotoxic and mitogenic to ovarian surface epithelial cells. In contrast, pregnancy-equivalent levels progesterone are highly effective as apoptosis inducers for ovarian surface epithelium and ovarian carcinoma cells [108]. Treatment with BPA, nonylphenol, octylphenol and methoxychlor for 24 h resulted in an increase of cell proliferation by increasing the ERE activity. The increase of cell proliferation and activation of ERE were reversed in the presence of an estrogen receptor antagonist, ICI 182780 [109]. But how they act?
Methoxychlor [MXC; 1,1,1-trichlor-2,2-bis(4-methoxyphenyl) ethane], for instance, is an organochlorine pesticide that has been primarily used since DDT was banned. In addition, triclosan is used as a common component of soaps, deodorants, tooth-pastes, and other hygiene products at concentrations up to 0.3%. Methoxychlor and triclosan stimulates ovarian cancer growth by regulating cell cycle and apoptosis-related genes via an estrogen receptor-dependent pathway [110]. The methoxychlor metabolite, 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE). Genistein and BPA stimulated both cell proliferation and induction of stromal cell derived factor 1 (CXCL12) mRNA and protein, in a manner comparable to estradiol, via their ability to activate the ER and displaying mitogenic activities in ovarian cancer cells [111].
Το our notice, while most cells undergo neoplastic transformation, including germ cells, granulose, and stromal cells, approximately 90% of tumors are derived from the ovarian surface epithelium. Similar to endometrial cancer, hormonal factors such as estrogen and xenoestrogens, have been linked to ovarian cancer. The development of appropriate biomarkers of cumulative exposure, and their measurement at developmental points where exposure is critical, is required to test the environmental estrogen hypothesis [112,113].
Conclusion
Recently, much evidence has accumulated showing that the environmental 'estrogens' can affect human and animal population. The issue involved is not the possible occurrence of short-term (maldevelopment) effects but the possible long term effects i.e., endometriosis, reproductive dysfunction or dysplastic changes that in humans may not manifest themselves until adolescence. Prospective studies as to the effects of endocrine disruptors are needed in order to allow potential hazards to ecosystem health, including humans, to be estimated.
References
- Bergeron JM, Crews D, McLachlan JA(1994) PCBs as environmental estrogens:Turtle sex determination as a biomarker of environmental contamination. Environ Health Perspect 102:780-781.
- Sager DB, Shih-Schroeder W, Girard D (1987) Effect of early postnatal exposure to polychlorinated biphenyls (PCBs) on fertility in male rats. Bull Environ ContamToxicol 38: 946-953.
- Dieringer CS, Lamartiniere CA, Schiller CM, Lucier GW (1979) Altered ontogeny of hepatic steroid-metabolizing enzymes by pure polychlorinated biphenyl congeners. BiochemPharmacol 28: 2511-2514.
- Woodward AR, Percival HF, Jennings ML and Moore CT(1993) Low clutch viability of American alligators on Lake Apopka. FlaSci 56:52-63.
- Guillette LJ, Gross TS, Masson GR, Matter JM, Percival F, et al.(1994) Developmental abnormalities of the gonad and abnormal sex hormone concentrations in juvenille alligators from contaminated and control lakes in Florida. Environ Health Perspect 102: 681-688.
- Feuer EJ, Wun LM (1992) How much of the recent rise in breast cancer incidence can be explained by increases in mammography utilization? A dynamic population model approach. Am J Epidemiol 136: 1423-1436.
- Henderson BE, Ross RK, Pike MC (1993) Hormonal chemoprevention of cancer in women. Science 259: 633-638.
- Wolff MS, Toniolo PG, Lee EW, Rivera M, Dubin N (1993) Blood levels of organochlorine residues and risk of breast cancer. J Natl Cancer Inst 85: 648-652.
- Davis DL, Bradlow HL, Wolff M, Woodruff T, Hoel DG, et al.(1993) Medical hypothesis. Xenoestrogens are preventable causes of breast cancer. Environ Health Perspect 101: 372-377.
- Wheeler JM (1992) Epidemiology and prevalence of endometriosis. InfertilReprod Med Clin 3: 545-549.
- Rier SE, Martin DC, Bowman RE, Dmowski WP, Becker L(1993) Endometriosis in rhesus monkeys (Macacamulatta) following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p- dioxin. Fund ApplToxicol 21: 433-441.
- Safe S, Astroff B, Harris M, Zacharewski T, Dickerson R, et al. (1991) 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and related compounds as antioestrogens: characterization and mechanism of action. PharmacolToxicol 69: 400-409.
- Bois FY, Eskenazi B (1994) Possible risk of endometriosis for Seveso, Italy, residents: an assessment of exposure to dioxin. Environ Health Perspect 102: 476-477.
- Damstra T, Barlow S, Bergman A, Kavlock R, Der Kraak Van (2002) WHO. Global Assessment of the State-of-the-Science of Endocrine Disruptors. WHO.
- MantovaniA (2002) Hazard identification and risk assessment of endocrine disrupting chemicals with regard to developmental effects. Toxicol 181-182: 367-70.
- Hertz R (1985)Theestrogen problem: Retrospect and prospect. In: Estrogen in the Environment II: Influences on Development.pp: 1-11.
- Ciccone MM, Scicchitano P, Gesualdo M, Fornarelli F, Pinto V, et al. (2013) Systemic vascular hemodynamic changes due to 17-ß-estradiol intranasal administration. J CardiovascPharmacolTher 18: 354-358.
- Ciccone MM, Cicinelli E, Giovanni A, Scicchitano P, Gesualdo M, et al.(2012) Ophthalmic artery vasodilation after intranasal estradiol use in postmenopausal women. J AtherosclerThromb 19:1061-1065.
- Clapauch R, Mecenas AS, Maranhão PA, Bouskela E (2012) Early postmenopausal women with cardiovascular risk factors improve microvascular dysfunction after acute estradol administration. Menopause 19: 672-679.
- Kurdoglu M, Yildirim M, Kurdoglu Z, Erdem A, Erdem M, et al. (2011) Cardiovascular risk assessment with oxidised LDL measurement in postmenopausal women receiving intranasal estrogen replacement therapy. GynecolEndocrinol 27: 551-557.
- Casanova G, Spritzer PM (2012) Effects of micronized progesterone added to non-oral estradiol on lipids and cardiovascular risk factors in early postmenopause: a clinical trial. Lipids Health Dis 11: 133.
- Stockard CR, Papanicolaou GN (1917)The existence of a typical oestrous cycle in the guinea pig; with a study of its histological and physiological changes. Am J Anat 22: 225-283.
- Long JA, Evans HM (1922) The oestrous cycle in the rat and its associated phenomena.
- Hadley M (2000). Endocrinology. Upper Saddle River, NJ: Prentice Hall.
- Di Cosmo A, Di Cristo C, Paolucci M (2001) Sex steroid hormone fluctuations and morphological changes of the reproductive system of the female of Octopus vulgaris throughout the annual cycle. J ExpZool 289: 33-47.
- Tarrant A, Atkinson S, and Atkinson M (1999)Estrone and estradiol-17 beta concentration in tissue of the scleractinian coral, Montiporaverrucosa. Comp BiochemPhysiol A MolIntegrPhysiol122:85-92.
- Pakdel F, Le Guellec C, Vaillant C, Gaelle Le Roux M, Valotaire Y (1989) Identification and estrogen induction of two estrogen receptors (ER) messenger ribonucleic acids in the rainbow trout liver: Sequence homology with other ERs. MolEndocrinol 3:44-51.
- White R, Jobling S, Hoare SA, Sumpter JP, Parker MG (1994)Environmentally persistent alkylphenolic compounds are estrogenic. Endocrinol 135:175-182.
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, et al. (1997) Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science 277: 1508-1510.
- Kuiper GGJM, Carlsson B, Grandien K, Enmark E, Haggblad J, et al. (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinol 138:863-870.
- vomSaal FS (1995) Environmental estrogenic chemicals. Their impact on embryonic development. Hume Ecol Risk Assess 1:3-15.
- Katzenellenbogen JA, O' Malley BW, Katzenellbogen BS (1996) Tripartite steroid hormone receptor pharmacology:Interaction with multiple effector sites as a basis for the cell- and promoter- specific action of these hormones. MolEndocrinol 10:119-131.
- Fenton SE (2006) Endocrine-disrupting compounds and mammary gland development: early exposure and later life consequences. Endocrinology 147:18-24.
- Villeneuve S, Févotte J, Anger A, Truong T, Lamkarkach F, et al. (2011) Breast cancer risk by occupation and industry: analysis of the CECILE study, a population-based case–control study in France. Am J Ind Med 54: 499-509.
- Labrèche F, Goldberg MS, Valois MF, Nadon L (2010) Postmenopausal breast cancer and occupational exposures. Occup Environ Med 67: 263-269.
- Brophy JT, Keith MM, Gorey KM, Laukkanen E, Hellyer D et al. (2002) Occupational histories of cancer patients in a Canadian cancer treatment centre and the generated hypothesis regarding breast cancer and farming. Int J Occup Environ Health 8: 346-353.
- Snedeker SM (2006) Chemical exposures in the workplace: effect on breast cancer risk among women. AAOHN J 54: 270-279.
- Soto AM, Brisken C, Schaeberle C, Sonnenschein C (2013) Does cancer start in the womb? altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J Mammary Gland BiolNeoplasia 18: 199-208.
- Birnbaum LS, Fenton SE (2003) Cancer and developmental exposure to endocrine disruptors. Environ Health Perspect 111: 389-394.
- denHond ED, Roels HA, Hoppenbrouwers K, Nawrot T, Thijs L, et al. (2002)Sexual maturation in relation to polychlorinated aromatic hydrocarbons: Sharpe and Skakkebaek’s hypothesis revisted. Environ Health Perspect 110:771–776.
- Miret N, Pontillo C, Ventura C, Carozzo A, Chiappini F, et al. (2016) Hexachlorobenzene modulates the crosstalk between the aryl hydrocarbon receptor and transforming growth factor-ß1 signaling, enhancing human breast cancer cell migration and invasion. Toxicology 366-367:20-31.
- Papoutsis AJ, Selmin OI, Borg JL, Romagnolo DF(2015) Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: preventive effects of resveratrol. MolCarcinog54:261-269.
- Romagnolo, Daniels KD, Grunwald JT, Ramos SA, et al. (2016) Epigenetics of breast cancer: Modifying role of environmental and bioactive food compounds. MolNutr Food Res 60: 1310-1329.
- Papoutsis AJ, Lamore SD, Wondrak GT, Selmin OI, Romagnolo DF (2010) Resveratrol prevents epigenetic silencing of BRCA-1 by the aromatic hydrocarbon receptor in human breast cancer cells. J Nutr 140:1607-1614.
- Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF (2009) Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3'-diindolylmethane in breast cancer cells. J Nutr 139:26-32.
- Parada H Jr, Wolff MS, Engel LS, Eng SM, Khankari NK, et al. (2016) Polychlorinated biphenyls and their association with survival following breast cancer. Eur J Cancer 56: 21-30.
- Parada H Jr, Wolff MS, Engel LS, White AJ, Eng SM, et al. (2016) Organochlorine insecticides DDT and chlordane in relation to survival following breast cancer. Int J Cancer 138: 565-575.
- Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W et al. 2006. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev 15:1509–1514.
- Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, et al. (2011) Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med 365: 1304-1314.
- Fenton SE, Birnbaum LS (2015) Timing of Environmental Exposures as a Critical Element in Breast Cancer Risk. J ClinEndocrinolMetab 100: 3245-3250.
- Bokhman JV (1983) Twopathogenetic types of endometrial carcinoma. GynecolOncol 15: 10-17.
- Sherman ME, Bur ME, Kurman RJ (1995) p53 in endometrial cancer and its putative precursors: evidence for diverse pathways of tumorigenesis. Hum Pathol 26: 1268-1274.
- Hale GE, Hughes CL, Cline JM (2002) Endometrial cancer: hormonal factors, the perimenopausal "window of risk," and isoflavones. J ClinEndocrinolMetab 87: 3-15.
- Misichronis G, Georgopoulos NA, Marioli DJ, Armeni AK, Katsikis I, et al. (2012) The influence of obesity on androstenedione to testosterone ratio in women with polycystic ovary syndrome (PCOS) and hyperandrogenemia. GynecolEndocrinol 28: 249-252.
- Visvanathan K, Vang R, Shaw P, Gross A, Soslow R, Parkash V et al. (2011) Diagnosis of serous tubal intraepithelial carcinoma based on morphologic and immunohistochemical features: a reproducibility study. Am J SurgPathol 35:1766-1775.
- Miller C, Degenhardt K, Sassoon DA (1998) Fetal exposure to DES results in de-regulation of Wnt7a during uterine morphogenesis. Nat Genet 20: 228-230.
- Newbold RR, Hanson RB, Jefferson WN, Bullock BC, Haseman J, et al.(1998) Increased tumors but uncompromised fertility in the female descendants of mice exposed developmentally to diethylstilbestrol. Carcinogenesis 19:1655-1663.
- Cline JM, WoodCE (2009) Estrogen/isoflavone interactions in cynomolgus macaques (Macacafascicularis). Am J Primatol 71:722-731.
- Liao CK, Rosenblatt KA, Schwartz SM, Weiss NS (2003) Endometrial cancer in Asian migrants to the United States and their descendants. Cancer Causes Control 14: 357-360.
- Women's Health Initiative Steering Committee (2004) Effects of conjugated equine estrogen in postmenopausal women with hysterectomy. Journal of the American Medical Association 291:1701-1712.
- Writing Group for the Women's Health Initiative Investigators (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Journal of the American Medical Association 288:321-333.
- Horn-Ross PL, John EM, Canchola AJ, Stewart SL, Lee MM (2003) Phytoestrogen intake and endometrial cancer risk. J Natl Cancer Inst 95: 1158-1164.
- Xu WH, Zheng W, Xiang YB, Ruan ZX, Cheng JR, et al. (2004) Soya food intake and risk of endometrial cancer among Chinese women in Shanghai: population based case-control study. BMJ 328:1285-1290.
- Goodman MT, Wilkens LR, Hankin JH, Lyu LC, Wu AH, et al. (1997) Association of soy and fiber consumption with the risk of endometrial cancer. Am J Epidemiol 146: 294-306.
- Buchanan DL, Sato T, Peterson RE, Cooke PS (2000).Antiestrogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in mouse uterus: critical role of the aryl hydrocarbon receptor in stromal tissue. ToxicolSci 57:302-311.
- Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, et al. (1998) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139:4252-4263.
- Akesson A, Julin B, Wolk A (2008) Long-term Dietary Cadmium Intake and Postmenopausal Endometrial Cancer Incidence: A Population-Based Prospective Cohort Study. Cancer Res 68:6435-6441.
- Hiroi H, Tsutsumi O, Takeuchi T, Momoeda M, Ikezuki Y, et al. (2004) Differences in serum bisphenol a concentrations in premenopausal normal women and women with endometrial hyperplasia. Endocr J 51: 595-600.
- Newbold RR, Padilla-Banks E, Jefferson WN (2006) Adverse effects of the model environmental estrogendiethylstilbestrol are transmitted to subsequent generations. Endocrinology 147:11-17.
- Newbold RR, Jefferson WN, Grissom SF, Padilla-Banks E, Snyder RJ, et al. (2007).Developmental exposure to diethylstilbestrol alters uterine gene expression that may be associated with uterine neoplasia later in life. MolCarcinog 46:783-796.
- Davis VL, Newbold RR, Couse JF, Rea SL, Gallagher KM, et al. (2012) Expression of a dominant negative estrogen receptor alpha variant in transgenic mice accelerates uterine cancer induced by the potent estrogendiethylstilbestrol. ReprodToxicol 34:512-521.
- Sturgeon SR, Brock JW, Potischman N, Needham LL, Rothman N, et al. (1998) Serum concentrations of organochlorine compounds and endometrial cancer risk (United States) Cancer Causes Control 9:417-424.
- Wernli KJ, Ray RM, Gao DL, Fitzgibbons ED, Camp JE, et al. (2008) Occupational risk factors for endometrial cancer among textile workers in Shanghai, China. Am J Ind Med 51: 673-679.
- Shen W, Han W, Li Y, Meng Z, Cai L, et al. (2016) Development of chemical isotope labeling liquid chromatography mass spectrometry for silkworm hemolymph metabolomics. Anal ChimActa 942: 1-11.
- Giudice LC, Kao LC (2004) Endometriosis. Lancet 364: 1789-1799.
- Eskenazi B, Warner ML (1997) Epidemiology of endometriosis. ObstetGynecolClin North Am 24: 235-258.
- Olive DL1, Schwartz LB (1993) Endometriosis. N Engl J Med 328: 1759-1769.
- Huhtinen K, Ståhle M, Perheentupa A, Poutanen M (2012) Estrogen biosynthesis and signaling in endometriosis. Mol Cell Endocrinol 358: 146-154.
- Parente Barbosa C, Bentes De Souza AM, Bianco B, Christofolini DM (2011) The effect of hormones on endometriosis development. Minerva Ginecol 63: 375-386.
- Bulun SE, Cheng YH, Pavone ME, Xue Q, Attar E, et al. (2010) Estrogen receptor-beta, estrogen receptor-alpha, and progesterone resistance in endometriosis. SeminReprod Med 28: 36-43.
- Rier S, Foster WG (2003) Environmental dioxins and endometriosis. SeminReprod Med 21: 145-154.
- Louis GM, Weiner JM, Whitcomb BW, Sperrazza R, Schisterman EF, et al. (2005) Environmental PCB exposure and risk of endometriosis. Hum Reprod 20: 279-285.
- Tsutsumi O (2005) Assessment of human contamination of estrogenic endocrine-disrupting chemicals and their risk for human reproduction. J Steroid BiochemMolBiol 93:325-330.
- Heilier JF, Nackers F, Verougstraete V, Tonglet R, Lison D, et al. (2005) Increased dioxin-like compounds in the serum of women with peritoneal endometriosis and deep endometriotic (adenomyotic) nodules. FertilSteril 84:305-312.
- Missmer SA, Hankinson SE, Spiegelman D, Barbieri RL, Michels KB, et al. (2004) In utero exposures and the incidence of endometriosis. FertilSteril 82: 1501-1508.
- Signorile PG, Spugnini EP, Mita L, Mellone P, D'Avino A, et al. (2010) Pre-natal exposure of mice to bisphenol A elicits an endometriosis-like phenotype in female offspring. Gen Comp Endocrinol 168: 318-325.
- Herington JL, Bruner-Tran KL, Lucas JA, Osteen KG (2011) Immune interactions in endometriosis. Expert Rev ClinImmunol 7: 611-626.
- Bruner-Tran KL, Rier SE, Eisenberg E, Osteen KG (1999) The potential role of environmental toxins in the pathophysiology of endometriosis. GynecolObstet Invest 48 Suppl 1: 45-56.
- Bruner-Tran KL, Ding T, Osteen KG (2010) Dioxin and endometrial progesterone resistance. SeminReprod Med 28: 59-68.
- Yu J, Wang Y, Zhou WH, Wang L, He YY, et al. (2008) Combination of estrogen and dioxin is involved in the pathogenesis of endometriosis by promoting chemokine secretion and invasion of endometrial stromal cells. Hum Reprod 23: 1614-1626.
- Nayyar T, Bruner-Tran KL, Piestrzeniewicz-Ulanska D, Osteen KG (2007) Developmental exposure of mice to TCDD elicits a similar uterine phenotype in adult animals as observed in women with endometriosis. ReprodToxicol 23: 326-336.
- Cai LY, Izumi S, Suzuki T, Goya K, Nakamura E, et al. (2011) Dioxins in ascites and serum of women with endometriosis: a pilot study. Hum Reprod 26: 117-126.
- Na YJ, Yang SH, Baek DW, Lee DH, Kim KH, et al. (2006) Effects of peritoneal fluid from endometriosis patients on the release of vascular endothelial growth factor by neutrophils and monocytes. Hum Reprod 21: 1846-1855.
- Koninckx PR, Kennedy SH, Barlow DH (1999) Pathogenesis of endometriosis: the role of peritoneal fluid. GynecolObstet Invest 1: 23-33.
- Heilier JF, Donnez J, Lison D (2008) Organochlorines and endometriosis: a mini-review. Chemosphere 71: 203-210.
- Anger DL, Foster WG (2008) The link between environmental toxicant exposure and endometriosis. Front Biosci 13: 1578-1593.
- Heindel JJ (2006) Role of exposure to environmental chemicals in the developmental basis of reproductive disease and dysfunction. SeminReprod Med 24: 168-177.
- Crain DA, Janssen SJ, Edwards TM, Heindel J, Ho S-M, et al. (2008) Female reproductive disorders: the roles of endocrine-disrupting compounds and developmental timing. FertilSteril 90:911-940.
- Sinaii N, Cleary SD, Ballweg ML, Nieman LK and Stratton P(2002). High rates of autoimmune and endocrine disorders, fibromyalgia, chronic fatigue syndrome and atopic diseases among women with endometriosis. A survey analysis. Hum Reprod 17: 2715–2724.
- Brinton LA, Gridley G, Persson I, Baron J, Bergqvist A (1997) Cancer risk after a hospital discharge diagnosis of endometriosis. Am J ObstetGynecol 176: 572-579.
- Obata K, Hoshiai H (2000) Common genetic changes between endometriosis and ovarian cancer. GynecolObstet Invest 1: 39-43.
- Chen CJ (2004) Reproductive Factors and Risk of Ovarian Cancer – Are Controls Really Controls?
- Strauss B (1995) Psychosomatic aspects of endometriosis. GeburtshilfeFrauenheilkd 55: M20-22.
- Strauss B, Didzus A, Speidel H (1992) A study of the psychosomatic aspects of endometriosis. PsychotherPsychosom Med Psychol 42: 242-252.
- Cunat S, Hoffmann P, Pujol P (2004) Estrogens and epithelial ovarian cancer. GynecolOncol 94: 25-32.
- Grow DR (2002) Metabolism of endogenous and exogenous reproductive hormones. ObstetGynecolClin North Am 29: 425-436.
- Rochefort H, Chalbos D, Cunat S, Lucas A, Platet N, et al. (2001) Estrogen regulated proteases and antiproteases in ovarian and breast cancer cells. J Steroid BiochemMolBiol 76: 119-124.
- Ho SM (2003) Estrogen, progesterone and epithelial ovarian cancer. ReprodBiolEndocrinol 1: 73.
- Park SH, Kim KY, An BS, Choi JH, Jeung EB, et al. (2009) Cell growth of ovarian cancer cells is stimulated by xenoestrogens through an estrogen-dependent pathway, but their stimulation of cell growth appears not to be involved in the activation of the mitogen-activated protein kinases ERK-1 and p38. J ReprodDev 55: 23-29.
- Kim JY, Yi BR, Go RE, Hwang KA, Nam KH, et al.(2014).= Methoxychlor and triclosan stimulates ovarian cancer growth by regulating cell cycle- and apoptosis-related genes via an estrogen receptor-dependent pathway. Environ ToxicolPharmacol 37:1264-1274.
- Hall JM, Korach KS (2013) Endocrine disrupting chemicals promote the growth of ovarian cancer cells via the ER-CXCL12-CXCR4 signaling axis. MolCarcinog 52: 715-725.
- Soto AM, Fernandez MF, Luizzi MF, OlesKarasko AS, Sonnenschein C (1997) Developing a marker of exposure to xenoestrogen mixtures in human serum. Environ Health Perspect 3: 647-654.
- Bencko V (2001) Human exposure to endocrine disrupters: carcinogenic risk assessment. Folia HistochemCytobiol 2: 24-25.

