Keywords
Incontinentia pigmenti; Atypical presentation; Genetic diseases
Case Report
10-day-old Caucasian female presented with her family to the emergency department for constipation. Dermatology was subsequently consulted for 7-day history of worsening rash. The mother had a normal pregnancy with prenatal care and a full term spontaneous vaginal delivery without complication. The patient was discharged from the hospital at Day 2 of life. Patient’s mother reports rash started as a few red bumps on her legs that spread to her trunk, upper extremities, and scalp. The rash did not seem to be symptomatic.
Physical exam was significant for firm pink papules and plaques over the trunk, arms, legs, and scalp along with scattered areas of fine desquamation on patient’s arms and abdomen (Figure 1). Patient had no palpable lymphadenopathy. We performed a 4MM punch biopsy on the patient’s right lateral thigh. Differential diagnosis at the time of biopsy was a depositional or fibrosing disorder versus subcutaneous fat necrosis of the newborn. Histopathologic examination demonstrated spongiosis with numerous eosinophils, and rare dyskeratotic cells within the epidermis consistent with Incontinentia Pigmenti (Figures 2, 3, and 4).
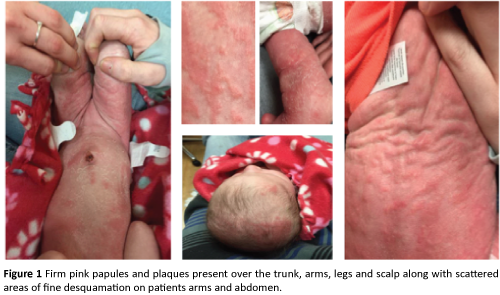
Figure 1: Firm pink papules and plaques present over the trunk, arms, legs and scalp along with scattered areas of fine desquamation on patients arms and abdomen.
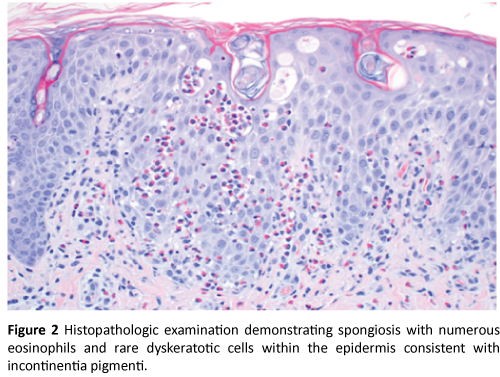
Figure 2: Histopathologic examination demonstrating spongiosis with numerous eosinophils and rare dyskeratotic cells within the epidermis consistent with incontinentia pigmenti.
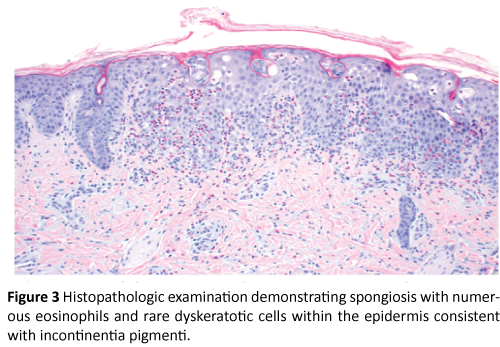
Figure 3: Histopathologic examination demonstrating spongiosis with numerous eosinophils and rare dyskeratotic cells within the epidermis consistent with incontinentia pigmenti.
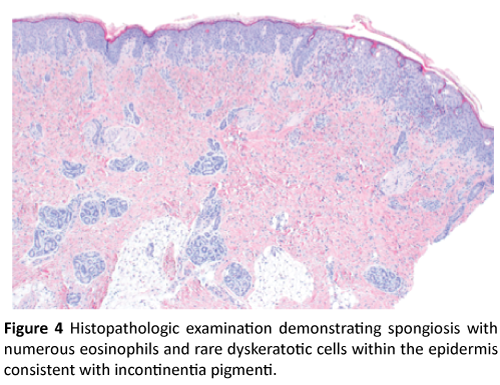
Figure 4: Histopathologic examination demonstrating spongiosis with numerous eosinophils and rare dyskeratotic cells within the epidermis consistent with incontinentia pigmenti.
Patient and family presented to the outpatient dermatology clinic for scheduled follow-up at day 14 of life to discuss biopsy results and re-evaluate rash. Since last evaluation, mother reported change in patient’s rash with more diffuse involvement and blisters forming on her arms and legs. They denied any known family history of skin disease or genetic disorders. Physical examination was significant for erythematous and serous-fluid filled vesicles following Blaschko lines of bilateral upper and lower extremities, yellow/white crusting of vertex scalp and mild diffuse desquamation of trunk and extremities (Figure 5). Patient’s family was counseled regarding the diagnosis and treatment plans. Patient was sent to be evaluated by Ophthalmology the same day and a consult was placed for evaluation by Genetics.
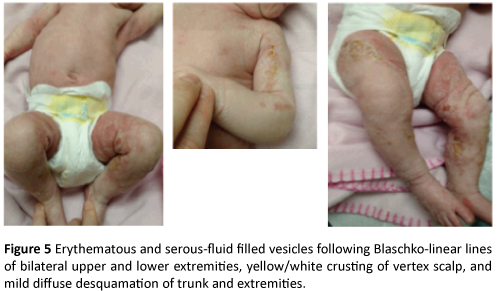
Figure 5: Erythematous and serous-fluid filled vesicles following Blaschko-linear lines of bilateral upper and lower extremities, yellow/white crusting of vertex scalp, and mild diffuse desquamation of trunk and extremities.
Discussion
Background information
Incontinentia pigmenti (IP) is an X-linked dominant disorder, also known as Bloch-Sulzberger Syndrome. The estimated prevalence of IP is 0.2/100,000 [1] with more than 700 documented cases worldwide [2]. Several organ systems are affected, including the skin, teeth, eyes, and central nervous system. The majority of IP patients are female, as it is commonly lethal in male fetuses. Diagnosis is typically made by the distinct dermatologic manifestations presenting in infancy, occurring in four stages: vesicular, verrucous, hyperpigmentation, and hypopigmentation/ atrophy [1].
Pathophysiology
The underlying pathophysiology of IP relates to the inhibitor of kappa B kinase gamma gene (IKBKG), previously known as NEMO, or nuclear factor kB essential modulator [1] on locus Xq28. This gene is 23-kb and has 10 exons, with over 80% of cases involving a deletion of exons 4-10. This deletion affects the normal function of nuclear factor kB, which is a transcription factor for regulation of immunoglobulin gene expression in B lymphocytes, genotoxic stress, inflammatory reactions, and cell adhesions, as well as protecting against apoptosis induced by tumor necrosis factor [2].
Although the majority of cases are caused by the same deletion, IP has variable phenotypic expression due to X-chromosome inactivation. Over 53 mutations have been reported, with 65% of mutations being de novo [1]. In a data report of IP patients from 2000-2013, it was found that 75.6% of cases were sporadic and 23.4% were familial, with 75.1% involving the NEMOdel4-10 mutation, 22.1% involving an IKBKG/NEMO point mutation, 3.6% with an extended deletion in the corresponding locus, and 4.7% with no known mutation in the IP locus [3]. There are reports of 120 cases in males, most of whom have Klinefelter’s Syndrome (47, XXY), somatic mosaicism, or hypomorphic alleles, contributing to their survival [2].
Clinical manifestations
The most distinctive clinical features of IP are the four stages of dermatologic lesions that typically present in infancy. Stage I is known as the vesicular stage, characterized by erythema and vesicles on the torso, extremities, and scalp, occurring in 90% of IP patients. Approximately 92% of patients present in Stage I during their first two weeks of life [2], however this stage can remain until four months of age and can recur with febrile illnesses. In a clinical study involving 20 patients with IP, 50% of patients also presented with peripheral eosinophilia [4]. Stage II is characterized by verrucous papules and plaques, with a distribution that is not dependent on Stage I lesions. This stage occurs in approximately 70% of patients, usually in the first two to six weeks of life [2]. Stage III is characterized by hyperpigmentation along the lines of Blaschko, involving the torso, extremities, nipples, axillae, and groin. These whorls and streaks of brown to gray pigmentation will occur in approximately 98% of IP patients at 12-26 weeks of age [2]. Unlike Stages I and II, Stage III can persist through adulthood, but typically resolves at puberty. There are reported cases with an initial Stage III presentation in a neonate, however in these cases, Stage I and II are thought to have occurred in utero [2]. Finally, Stage IV is characterized by hypopigmentation and atrophy. Lesions can present as pale, hairless, atrophic plaques or non-tanning hypopigmented areas without atrophy, typically involving the extremities.
Additional organ systems involved in IP include the hair, teeth, eyes, and central nervous system. Approximately 50% of patients have anomalies of the hair, with the most common presentation being vertex alopecia [1], followed by reports of wiry and coarse hair. Nail anomalies usually present at puberty, with findings including ridging, pitting, dystrophy, and subungual or periungual tumors. These manifestations can present in up to 64% of patients [3]. Approximately 80% of IP patients are thought to have dental manifestations, most commonly partial anodontia, followed by pegged and conical teeth and late dentition [2]. The most serious manifestations of IP, however, involve the eyes and central nervous system. Approximately 35% of patients have ocular abnormalities, which can be divided into retinal and nonretinal manifestations. Retinal findings include foveal hypoplasia, mottled or hypopigmented retinal pigment epithelium, avascular retina, neovascularization, vitreous hemorrhages, fibrovascular proliferation, and retinal detachment. Non-retinal findings can include strabismus, optic nerve atrophy, conjunctival pigmentation, iris hypoplasia, nystagmus, and uveitis [2]. The most common ocular manifestations are vision defects [3]. Finally, 13-35% of IP patients are affected by central nervous system abnormalities. The most common manifestation is seizures (40%), followed by mental retardation (29.9%), motor impairment (16.5%), and microcephaly (11.3%) [3].
Atypical presentations reported in the literature
Here we report a patient who presented with papules and plaques followed by the typical progression of skin lesions – an atypical presentation not previously found in the literature. Because of the phenotypic variability of IP, there are several reports of unusual presentations.
Chang, et al conducted a 12-year study on four patients diagnosed with IP – three females and one male. Two females presented in Stage I, one female presented in Stage III, and the male infant presented presumably in Stage I, with vesicular lesions only involving one limb. The female infant who presented in Stage III had concurrent left foot deformity and isolated nail dystrophy, one of the first presentations involving a limb anomaly [4]. Only one of these four patients was found to have a family history of IP. It was subsequently found in an additional study that 8 out of 9 surviving males presented with lesions only involving one extremity or one side of the body [5].
In a 2005 case report, a female infant presented with persistent pulmonary hypertension of the newborn as well as classic Stage I lesions of IP with progression to Stage IV hypopigmentation. She had abnormal MRI findings involving polymicrogyria with cortical dysplasia and retinal hemorrhages. She subsequently developed fatal pulmonary hemorrhage. This patient was confirmed to have a NEMOdel4-10 and is one of only 2 IP patients who have presented with PPHN that was unresponsive to medical treatment, leading to the hypothesis that the NF-kB signaling pathway could have a relation to vasculopathies, as seen in the CNS and retinal vessels of IP patients [6]. This theory is supported by an additional case of a surviving male infant who died from severe hemorrhage 24 hours after birth. His mother subsequently became pregnant with another affected male child, at which point she terminated the pregnancy and suffered from severe bleeding [7].
In a 2016 clinical study of 20 patients with IP, four patients presented with anomalies not previously seen in IP patients including Wilm’s tumor, congenital hypothyroidism, Still’s disease with macrophage activation syndrome, and myasthenia gravis. These findings support the proposed pathophysiology involving the NF-kB signaling pathway, which is associated with inflammatory and immune processes [4]. This theory is also supported by a case report involving a male infant with IP and immune dysfunction who developed multiple infections due to poor lymphocyte function and low IgG levels, heat intolerance with hyperthermia, anhidrosis, eczema, weight loss, abdominal pains, hepatosplenomegaly, and contraction of mycobacterium avium intracellulare [7].
Finally, there is a case report of an abnormal presentation involving a patient who presented with hyperpigmented plaques at birth, followed by a pruritic, flat, hyperpigmented whorled rash along Blaschko lines at age 14 months. This patient’s workup showed increased plasma histamine and increased mast cells, which led to an initial diagnosis of Urticaria Pigmentosa, however her clinical presentation was subsequently determined to be consistent with IP [8].
Conclusion
In conclusion, Incontinentia Pigmenti is an X-linked dominant disorder caused by a NEMO deletion in the Xq28 locus with a wide phenotypic variability, occurring more commonly in females than males. As demonstrated by the several reports of atypical presentations, including our patient’s presentation with papules and plaques, there is much to be discovered in regards to atypical presentations and progression of dermatologic manifestations with relation to the varying mutations involving NF-kB. Based on this presentation, Incontinentia Pigmenti can be added to the differential diagnosis of infants presenting with papules and plaques, allowing for earlier diagnosis and follow-up of systemic manifestations.
References
- Minic Tripnic SD, Obradovic M (2014) Incontinentia Pigmenti Diagnostic Criteria Update. Clinical Genetics 85: 536-542.
- Alexander B, Paller A, Lawrence C (2002) Incontinentia Pigmenti: A Review and Update on the Molecular Basis of Pathophysiology. Journal of American Academy of Dermatology.
- Francesca F, Paciolla M, Conte MI, Pescatore A, Esposito E, et al. (2014) Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet Journal of Rare Diseases 9: 93.
- Poziomczyk P, Claudia S, Bonamigo RR, Santa Maria FD, Zen PR, et al. (2016) Clinical Study of 20 Patients with Incontinentia Pigmenti. International Journal of Dermatology 55: e87-e93.
- Jenn-Tzong C, Chiu PC, Chen YY, Wang HP, Hsieh KS (2008) Multiple Clinical Manifestations and Diagnostic Challenges of Incontinentia Pigmenti – 12 Years’ Experience in 1 Medical Center. Journal of Chinese Medical Association 71: 9.
- Sunit G, McNamara P, Rajguru M, Hellmann J (2005) Perinatal/Neontal Case Presentation: Unusual Neonatal Presentation of Incontinentia Pigmenti with Persistent Pulmonary Hypertension of the Newborn: A Case Report. Journal of Perinatology 25: 289-292.
- Aradhya S, Courtois G, Raijkovic A, Lewis RA, Levy M, et al. (2001) Atypical Forms of Incontinentia Pigmenti in Male Individuals Result from Mutations of a Cytosine Tract in Exon 10 of NEMO (IKK-gamma). American Journal of Human Genetics 68: 765-771.
- Donahue D, Erin C, Sonal R, Patel A. Case of Incontientia Pigmenti Masquerading As Urticaria Pigmentosa. White Memorial Medical Center, Los Angeles, CA. Journal of Allergy and Clinical Immunology 133: 2.