Keywords
DNA methylation; Histone modifications; Genome imprinting; Beckman Wiedeman syndrome; Russell Silver syndrome; lncRNAS; Xist
Introduction
Epigenetic mechanisms play a fundamental role in controlled development and gene expression in different types of cells of an organism, carrying the same genomic DNA sequence. These mechanisms control differences in the gene expression that are mitotically heritable although not altering the primary DNA sequence [1]. A large number of proteins write, read or erase particular epigenetic modifications and thus define where and when the transcriptional machinery can access the primary DNA sequence to drive normal growth and differentiation in the developing embryo along with the fetus. Different type of epigenetic marks work in concert to drive appropriate gene expression. These are DNA methylation at CpG dinucleotides, covalent modifications of histone proteins, noncoding RNA’s (ncRNA) along with other complementary mechanisms contributing to higher order chromatin organization, within the cell nucleus. There are two special examples e.g., chromosome inactivation and genome imprinting, which explains how important are the epigenetic mechanisms in regulating correct patterns of gene expression during early development chromosome inactivation basically is an example of dosage compensation in females leading to monoallelic expression of a huge number of X linked genes in female. Genome imprinting is a process in which special genes carrying epigenetic marks from parents of origin have the capacity for getting monoallelic parent of origin specific cell types at specific times of development. In germ cells in development as well as in embryo, there is genome wide reprogramming which is responsible for erasure as well as reestablishing of the correct epigenetic patterns. In contrast to these naturally occurring processes, the processes used in induced pluripotent stem cells from somatic cells are quite different [2], reviewed by Huang et al[3].
Changes in epigenetics can occur by different mechanisms and lead to infertility and imprinting disorders. Genetic as well as environmental factors impact genetic marks, which develop phenotypic differences varying from normal variation to human disease [4]. Both environmental factors e.g., starvation as well as artificial reproductive technologies (ART) have been shown to affect the epigenome of the embryo e.g., of the epigenetic changes which are associated with maternal starvation in fetal life can remain throughout adulthood, contributing to late onset disorders e.g., CVS disorders and type 2 diabetes mellitus [5-9].
Epigenetic marks
There is lot of crosstalk between various epigenetic marks like DNA methylation, histone modifications, NC RNA to regulate epigenome [10,11]. The ENCODE project, was a large collaborative one, which was developed to define all of the functional elements in the human genomes, got published recently having big datasets regarding histone modifications, transcription etc. These data point to both global as well as regional changes which overlap regarding epigenetic features, which in combination regulate gene expression [12].
DNA methylation
DNA methylation is one of the most studied mechanisms [13]. Methylation is associated with gene silencing through binding of methylation sensitive DNA binding proteins and or by interacting with various modifications of histone proteins, which modulate access of gene promoters to transcriptional machinery [14]. Basically DNA methylation in eukaryotic species involves the transfer of a methyl group to the cytosine of the CpG dinucleotide.
Histone modifications
Chromatin has a basic unit which is made up of an octamer of histone proteins, 2 each of H2A, H2B, H3 and H4, DNA wraps around this core which gives stability to the structure along with capacity to regulate gene expression. Each core histone within a nucleosome has a globular domain along with a very dynamic N-terminal tail extending from the globular domain. They have tails which can cause a number of post translational modifications, which induce acetylation, methylation, phosphorylation, ubiquitylation, sumoylation. ADP and ribolysation proteins isomerization, citrullization, butyration, propionylation, and glycosylation [15]. 11 histone post translational modifications, were analyzed in the ENCODE project data, which includes acetylation along with methylation which mark active as well as regressive chromatin, besides modifications which were associated with transcription. They identified different chromatin stages which induced an active, bimodal, and inactive, each of which has different functional properties [15]. Bimodal data in which a combination of active and repressive marks are there, in the chromatin, of the promoter region of the gene, helps in reprogramming changes in gene expression which might be expected during early development, when differentiation and specification occur [16].
Regulatory ncRNAs
Eukaryotic gene transcripts, upto75% of genomic DNA, roughly 3% of these transcripts encode for proteins, of which main are ncRNAs, which are classified based on their size and function [12,15,17]. They include small interfering RNA (Si RNA), micro RNA (miRNA) and long noncoding RNA (lncRNA), which have important roles in gene expression, regulation at various levels, like transcription, degradation of mRNA, splicing and translation [18]. Si RNA’s are double stranded RNA (dsRNA), which mediates posttranscriptional silencing, which is in part done by inducing heterochromatin to recruit histone deacetylase complexes [19]. MiRNA are class small 18-24 nucleotides in length [20-22].
Linc RNA, a subset of lncRNA shows high conservation across different species. They have been shown to guide chromatin modifying complexes to specific genomic loci establishment of and participating in cell types-specific epigenetic states. In embryonic development, especially by lncRNA regulated by the pluripotent transcription factors like OCT4 and NANOG facilitate cell lineage specific gene expression [23]. They also play an important role in development processes of X chromosomal inactivation and genome imprinting [24].
The Role of long noncoding RNA (lncRNA) in dosage compensation
In species with genetic sex determination such as XX female, XY male system in mammals and in the ZW female; ZZ male system in birds, males and females have a difference in sex chromosome-linked gene dosage which has resulted in the evolution of dosage compensation mechanism in mammals which is realized by the inactivation of one of the X chromosomes through coating by a lncRNA called Xist (Xinactive specific transcript). The 19 kilobase long transcript Xist is only transcribed from inactive X chromosome and coats hundreds of genes. Prior to inactivation an lncRNA that is antisense to Xist called Tsix is down regulated from one of the X chromosomes resulting in the Xist and inactivation of the X chromosome. On the active X, the maintenance expression of Tsix prevents the full-length Xist expression and X linked gene expression is unaffected (reviewed by Moran [25]). This phenomenon on dosage compensation in mammals is clearly regulated by lncRNAs. But in other groups such as birds there is no inactivation of one sex chromosome of the homogametic sex. However there is potential involvement of lncRNAs in dosage compensation in chicken as well. Chickens and other birds have a ZZ male, ZW female sex chromosome system.
The Z-linked transcription factor gene DMRT1 is thought to play a role in avian sex determination by directing testis development in Zz embryos. Overexpression of DMRT1 induces the male specific genes HEMGN, SOX9 and AMH (Figure 1) [26].
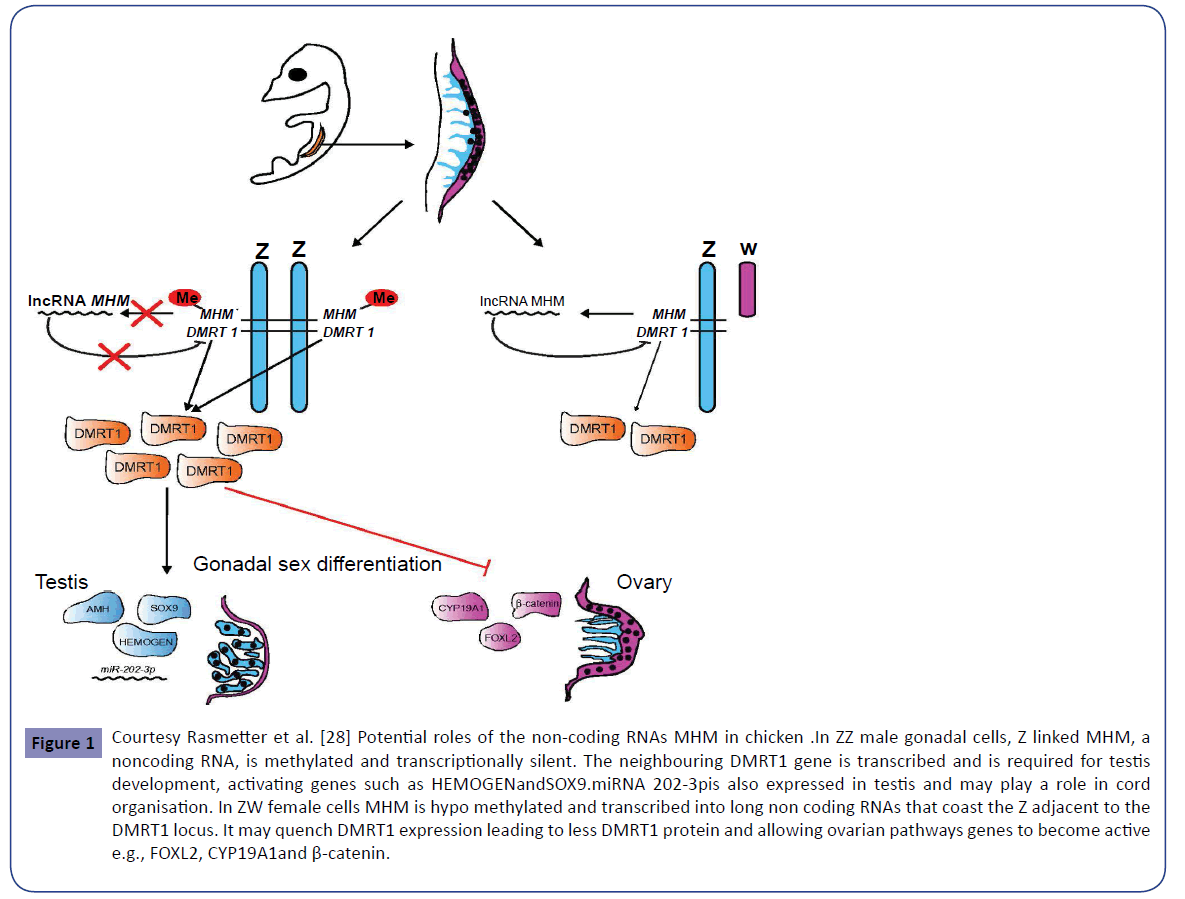
Figure 1: Courtesy Rasmetter et al. [28] Potential roles of the non-coding RNAs MHM in chicken .In ZZ male gonadal cells, Z linked MHM, a noncoding RNA, is methylated and transcriptionally silent. The neighbouring DMRT1 gene is transcribed and is required for testis development, activating genes such as HEMOGENandSOX9.miRNA 202-3pis also expressed in testis and may play a role in cord organisation. In ZW female cells MHM is hypo methylated and transcribed into long non coding RNAs that coast the Z adjacent to the DMRT1 locus. It may quench DMRT1 expression leading to less DMRT1 protein and allowing ovarian pathways genes to become active e.g., FOXL2, CYP19A1and β-catenin.
MHM is a 2.2 kilobase sequence absent in other birds and MHM is located within a region of Z chromosome which corresponds to hyper acetylation of histone H4 which is associated with increased gene expression or second hypothesis MHM may regulate by in male cells ZZ MHM is hyper methylated and transcriptionally silent, whereas in female cells Z which is hypo methylated and transcribed. Being near DMRT1, it is suggested it may influence to dampen DMRT1 in female cells, by MHM lncRNA coating the chromosome adjacent to DMRT locus, inducing local chromatin conformational changes which may interfere by TF binding [2]-7further reviewed by Rastetter et al.
Special Types of Epigenetic Regulation
XInactivation: This compensates for differences, in X linked genes, females silence most of one of their 2X Chromosome through a process known as X chromosome inactivation [29]. Embryo s having > 1XC (XXX, XXY) undergo rapid X chromosome inactivation at the blastocyst in the early embryogenesis [30]. X chromosome inactivation is controlled by a master switch locus called the X Inactivation Centre (XIC), which regulates in cis the expression of lincRNA gene XIST (Xinactive specific transcript) and also antisense transcript unit TSIX. In mouse the XIC senses the number of X C’s. This involves coating of all future inactive X chromosomes by the Xist RNA followed by polycromb repressor complex 2 reruitment and then addition of silencing chromatin marks such as histone H3 and H4 hypoacetylation. H3 lysine 27 methylation and DNA methylation at CpG rich promoters [31,32]. This process which is still incompletely understood restricts each diploid cell to one active copy of the X chromosome.
Genome Imprinting: This is an epigenetic process by which the male and the female germline impart specific marks or imprints, onto particular chromosomal regions [33]. With these certain chromosomal regions provide capacity for monoallelic parent of origin specific expression at certain times of development or in specific cell types. For a normal imprinted gene expression, number of specific paternal alleles needs contribution from alleles from both parents. Thus imprinted dysregulation results from changing the number or appropriation of paternal alleles. Extreme examples are where contribution occurs in conceptuses only from one parent of origin, either maternal or paternal. In mature cystic ovarian teratoma (MCT) occurring from meiotic errors during oocyte maturation (gynegenetic fetus) only maternally derived chromosomes are found. This causes the development of a cyst which contains tissues from all 3 embryonic germ layers. While in contrast in an androgenetic conceptus, carrying two paternal genomes and no maternal genome, fails to drive embryonic life, fetal development and results in hydatidiform mole (AnCHM) MCT and AnCHM display the serious biological consequences of uniparental inheritance [34], which demonstrates that both parental genomes are required for normal development of an embryo with the paternal genome being required for extraembryonic tissues and maternal genome being essential for embryonic development.
Autosomal genes get expressed from both paternal and maternal alleles whereas imprinted genes are expressed almost entirely from one allele, the maternal or the paternal in a specific manner being from parent of origin. Thus the imprinted gene gets expressed from the paternal allele when the maternal counterpart gets silenced or reversely it is the maternal allele which is expressed and paternal allele is silenced [35]. These Imprinted genes do not follow Mendelian genetics and one cannot predict parent of origin specificity for gene expression [36]. Roughly 100 genes have been reported as imprinted on the website https://igc.otago.ac.nz/home.html . These genes can play important pivotal roles in embryonic, placental, fetal growth along with neurodevelopment [36-38]. Imprinted genes cluster together forming imprinted domains, which have been found on C number 6, 7, 11, 14, 15 and 20 in human beings [38-40]. Imprinted genes get regulated by imprinting centres (ICs) present on these domains. Such ICs are characterized by differential methylated regions (DMRs). These DMRs carry parent of origin specific DNA methylation and histone modification marks. These cis acting DMRs along with Trans acting factors form the basis of the parent of origin specific gene expression of imprinted genes, e.g., are insulin like growth factor 2 [(IGF2/H19)-IC core regulates the paternal expression of IGF2, with the maternal expression of H19,2 genes located practically in the same imprinted domain 90 kilobase apart [41].
PGCs and Epigenetic Reprogramming
In PGCs there is erasure involving whole genome and reprogramming of epigenetic marks. Whether egg or sperm will develop from such early precursors is dictated by the sex of the developing embryo. In this sex specific differentiation there is parent of origin specific prints which get established in sperms and oocytes. In mice it has been seen that PGCs move towards genital ridge at day 8.5 of embryonic life [E8.5] and then reaching endpoint by E11.5 [42]. Between E11.5 and E13.5 at the site where future gonads develop is believed where genome wide demethylation of PGCs occur [43]. This methylation decreases globally with only 7% of CpGs remaining methylated, as compared to 70-80% embryonic stem cells and somatic cells [44]. Gene promoter specific for germ cells get methylated in early PGCs, and become demethylated and are expressed during reprogramming [45]. At this time as well PGCs of female embryo down regulate Xi RNA expression from the inactive X chromosome (Xi) [46,47], thus equivalent two X Cs may participate in meiosis for the female gamete production. Methylation in PGCs is established by Dnmt3a and Dnmt3L recruitment [48]. ZFP57 (a KRAB zinc finger protein) also plays important role in oocyte imprint establishment [39,49]. Timing of gamete development differs between male and female embryos. Epigenetic reprogramming of oocyte begins much later than in sperm. Beginning at puberty, is nearly complete in each oocyte at the time of ovulation.
Role of oocyte candidate transcriptomic factors
Global epigenetic analysis has shown that mammalian metaphase II oocytes have a greater capacity to epigenetically reprogramed somatic cell nuclei towards ESC like state [50-52]. Recently Zhou et al. demonstrated a T cell dependent immune response upon transplantation into a perfectly matched syngeneic mouse, a phenomenon which is not seen in syngeneic transplantation of ESC [53]. Hence it was considered maybe oocytes possess specific factors which are lacking in current factor based reprogramming approaches. With the suggestion by various authors on global genetic analysis that mammalian metaphase II oocytes may possess a higher capacity to epigenetically reprogram somatic cell nuclei toward an embryonic stem cell (ESC) line like state, Awe et al. proposed, based on the suggestions of Gurdon and Wilmut that the oocyte maybe involved in loosening somatic chromatin [54] and thereby providing the transcription regulatory apparatus access to repressed genes by which they would significantly increase epigenetic reprogramming [55].
To test this hypothesis they tested a list of candidate oocyte reprogramming factors (CORF) which are significantly expressed in metaphase II oocyte. Having focused on 2 different species in earlier studies unbiased global analysis of oocytes from 3 species (human, rhesus monkey, mouse) demonstrated 8 CORFs (ARID2, ASFTA, ASFTB, DPPA3, ING3, MSL3, HIFOO, and KDM6B ) having significant (p < 0.05, FC > 3)expression in oocytes of all 3 species, having well established roles in loosening/up chromatin structure. Besides that they identified additional CORFs which fit with their proposed chromatin opening fate transformation (COFT) model. ARID2, which plays a key role in activating gene expression through the PBAF chromatin remodelling complex [56]. ASF1A and ASF1S which are histone-remodelling chaperones that cooperate with chromatin assembly factor1 (CAF1), which plays a key role in remodelling chromatin in pluripotent embryonic cells [57,58]. BRDT which plays a role in the reorganization of acetylated chromatin in germ cells [59]. DPPA3 and DPPA5 which are pluripotency associated factors with DPPA3 in particular playing a known role in altering chromatin structure in oocytes [60,61]. Rps6ka5 which contributes to gene activation by histone phosphorylation [58]. TADA2L, a component of the ATAC complex which, has histone acetyl transferase (HAT) activity on histones H4 and H2A. ING3, a component of the NuA4 HAT x that is involved in transcriptional activation of select genes principally by acetylation of nucleosomal histones H4 and H2A. MLL3, which activates transcription through methylation of Lys-4 of Histone H3 and is essential in maintaining the haematopoietic stem cell state [62]. 8B). MSL3, a component of the MSL complex that is responsible for majority of histone H4 acetylation at Lys-16 which is implicate in the formation of more open chromation structure, specially by inhibiting the formation of the compact 30-nanometer-like fibers and impending the ability of chromatin to form crossfiber interactions [63]. NCOA3, a nuclear receptor coactivator that displays HAT activity [58]. HIFOO, the oocyte specific linker histones that has greater mobility than somatic histones and plays a key role in generating the increased instability of the embryonic chromatin structure following fertilization and somatic cell nuclear transfer [64] and KDM6B, a histone demethylase that specifically demethylases Lys-27 of Histone H3 here by prevents the formation of repressive chromatin through polycromb group (PcG) protein complex PRC1 building [65]. These CORFs may be able to augment both Takayashus and Shinya Yamannaki’s previous identified reprogramming factors (Oct4 or POU5F1), Sox2, Kruppel like factor 4 (KLF4), cMYC and potentially facilitate the removal of epigenetic memory in induced pluripotent stem cells and reduce the expression of immunogenicity of genes in iPSc derivatives, having applications in personalized PSC based therapeutic.
Male infertility and epigenetics
Male Infertility and Methylation: In DNA methylation there is methyl group addition to 5th position of cytosine ring (5 meC) in CpG dinucleotide from 5’adenosyl methionine; 3-5% of cytosine dinucleotide in mammalian genomic DNA, appear in the form of 5 meCP [66]. By definition CpG islands are sequences with an observed to expected ratio of CpG > 0.6 and with length > 500 bp [67]. They are not randomly distributed in the genome. CpG island promoters are mostly unmethylated e.g., housekeeping genes promoters, whereas CpG islands located in the promoter and/or in the regulatory regions of transposable elements are methylated, which involves the parasitic transposable and repetitive elements from replicating CpG promoters are frequently hypermethylated in somatic cells.
New data point to DNA methylation may occur in embryonic cells at cytosine residues in CpA or CpT residues [68,69]. During male germ development Sasaki et al. showed there was accumulation along with loss of asymmetric non CpG methylation. Still the bialleliic form of non CpG methylation is unclear [70]. Methylation getting established is necessary for proper spermatogenesis and sperm production [71].
It was shown in mouse germ cells that there is global demethylation–remethylation in which erasure of somatic cells patterns by denovo DNA methylation [66,72-75].
Demethylation occurs in PGCs between 8-13 days post coitum (dpc) of the developing embryo and erases all methylation marks [45,76,77]. Specialized remethylation occurs in spermatogonic and type 1 spermatocytes before they enter meiosis, thus all spermatozoa transmit correct paternal imprint [72].
Although methylation gets acquired mostly during fetal life complete levels of DNA methylation do not get achieved till the pachytene spermatocyte stage, thus postnatally and before meiosis [78]. Four imprinted loci are methylated in male germline only namely; Igf2/H19, Rasgrf1, Dlk1-Gtl-2, and Zdbf2.
Activities of DNMTs are absolutely essential for normal completion of spermatogenesis. Conditional Dnmt3a knockout (KO) in germ cell impairs spermatogenesis by germ cell apoptosis and is associated with methylation of paternally imprinted genes [48]. Meiotic arrest is induced in Dnmt3 LKO due to chromatin a synapsis. Dysregulation of methylation process of imprinted genes and repeated sequences is also observed. Having been studied in mice exhaustively in humans very few studies exist. Only in two studies there was methylation of H19DMRin spermatogonia and also the expected demethylated stage of MEST/PEG1 in spermatogonia and type1 spermatocytes [79,80]. In human beings it is thought that just like in mice these methylation markers get acquired before entry into meiosis. Though it is not clear whether it occurs during fetal life, perinatal period or the pubertal period. Studying relation between male infertility in humans with use of immunostaining for DNA methylation in a study, if there were high global methylation levels of ejaculated sperm DNA, it corresponded to higher pregnancy rates as compared to those having methylation defeats having infertility [81]. Using Methylight staining of 36 target genes using ilumina platform showed improperly increased levels of DNA methylation of imprinted genes along with repetitive elements in poor quality semen samples [82]. According to them increased methylation occurred due to an improper erasure of methylation marks in cases of oligo-astheno-teratozoospermia (OAT), rather than from denovo methylation following epigenetic reprogramming. They suggested that besides imprinted genes broad epigenetic defects were also seen with sperm defects.
Aston et al. utilizing Ilumina infinium human methylation 27 Bead chip assay showed that there was a genome wide altered DNA methylation pattern in man with whose semen parameters were defective [83], thus correlating global methylation levels of sperms with infertility. Marques studying imprinted genes comparing paternal methylated H19DMR and unmethylated MESTDMR in spermatozoa from fertile and infertile men. They found loss of methylation of H19DMR in men with OAT along with an association between H19DMR methylation decrease and decreased sperm count [84,85]. An abnormal methylation state of MEST DMR was also seen in oligozoospermic patients contrary to expectations. This was confirmed by Kobayashi et al. on various other imprinted genes which showed loss of methylation at H19 and GLT2DMRs, and improper methylation acquisition at PEG 1, LIT1, ZAC, PEG3, SNRPN loci in cases of moderate–severe oligozoozoospermia [86]. Also one found association between sperm abnormalities after ART with same imprinting errors reported [87,88]. Although techniques used for methylation study was bisulfate conversion and cloning sequencing techniques. This technique shows allele specific methylation state of but represents poorly the methylation status of samples being examined. With the use of combination of bisulfate conversion with pyro sequencing a qualitative analysis of methylation levels at each CpG position included in IGF2and H19DMR (47 different CpGs) in a population of both oligozoospermic as well as normozoospermic men [88]. They confirmed that the drastic loss of methylation restricted to IGF2DMR2 (mainly IGF2DMR2) as well as H19DMR correlated with the severity of oligozoospermia. From this data, it was suggested that there are few DMRs having a greater sensitivity to defects of methylation as compared to others. Probably different DNA compaction state of following sites, explained by Boissonas [88].
Various other groups confirmed errors of methylation (hypermethylation or demethylation) which were associated with OAT with use of various techniques [83,89-91]. Some groups studied methylation of promoter implicated in spermatogenesis. There was abnormal methylation of the DAZL promoter [92] and the CREM promoter [93,94], were shown in men having OAT methylation status of the promoter genes involved in methylene tetrahydrofolate reductase (MTHFR) gene promoter was studied [95]. This particular gene encodes an important enzyme of folate pathway, which maintains availability of methionine. Methionine can be converted toS-adenosylmethionine, the universal methyl donor for many substrates which includes DNA hyper methylation of MTHFR promoter was seen in 53% of non-obstructive zoospermia [96] and in some cases of idiopathic infertility [97]. Still there are a lot of unanswered questions regarding methylation errors. One possible explanation is that abnormal DNA configuration during spermatogenesis where histones and protamine play a role in the maintenance on methylation markers of male gametes [98].
Histone Modifications: For histone acetylation/removal, of acetyl group, one needs the enzymes histone acetyl transferase and decaetylase, respectively, to activate genes [99]. Chromatin relaxation occurs following histone acetylation and thus makes it more available for transcription factors while decetylation brings about gene silencing [99]. H3 & H4 lysine residues acetylation is high in male stem cells and gets removed during meiosis but there is reacytylation of H4K in elongated spermatids, which is a prerequisite for histone to protamine exchange [100]. Use of an HDAC inhibitor trichlorstane a caused a marked decrease in the number of spermatids and severe male infertility [101,102]. Methylation of H3 or H4 lysine residues can promote gene activation or repression is regulated by histone methyl transferases (HMT) or histone deacetylases (HDM), generally associated with gene expression, whereas H3K4 methylation is generally associated with gene expression, whereas H3K9 and H3K27 methylation is linked to gene silencing and heterochromatin. There is importance of establishment and removal of methylation markers, which is crucial for spermatogenesis. Mono, di and trimethylation modifications of H3K4, H3K9, and H3K27peaks and increases during meiosis, but the removal of H3K9 by the end of meiosis is a must for spermiogenesis onset. There are enzymes responsible for these modifications. In mice reduction of histone H3K4 methyl transferase MII2 activity=>marked decrease in number of spermatocytes by an apoptotic process=>developmental block in the differentiation of spermatocyte cycle [103]. Also loss of LSD1/KDM1 an H3K4HDM during meiosis gives rise to germ cell apoptosis and infertility [104,105]. Disruption of JHDM2A in mice causes total loss of TNP1 and P1 expression, with defective chromatin condensation and infertility [1]. JHDM2A is an H3K9HDM, has a targeted action during spermiogenesis. JHDM2A binds to the core promoter regions of TNP1 and P1 lead to induction of transcriptional activation of removal of H3K9 methylation. Crotonoyl methylation (C4H50) addition on lysine residues of all core histones leads to histone modifications known as crotonoylation. It makes sex chromosome but also gonosomes and confers gene resistance to transcription repressors [107]. Thus histone tail modifications play important role in spermatogenesis.
Role of histone to protamine transition
Transition from histone to protamine is an important step in regarding spermiogemesis, which is favoured by histone hyper acetylation [108]. The P1/P2 ratio normally is 0.08-1.2 [109] in humans. There is increased DNA fragmentation, suggesting that incorrect DNA compaction, is more exposed to DNA damage and more oxidative stress [110-113]. Alterations of P1/ P2ratio are very rare in fertile men but common in men with infertility [15,111,114]. Both increased and decreased P1/P2 ratio=>infertility and thus under expression of either of P1 and P2 are linked to male subfertility [115-118]. Brunner et al. by novel proteomic approach combining peptide based bottom up and intact protein top down mass spectrometry, identified epigenetic marks on histones and protamines in the mouse sperm. A total of 26 posttranslational modifications(PTMs)on specific residues of the core H2B, H3 & H4 and the linker histone h1 four of which had not been described previously in any tissue or cell line. They also found 11 novel PTMs on the protamine PRM1 and PRM2 and observed that they are present in specific combination on individual protamines. They concluded both histones and protamine carry multiple PTMs in the adult mouse sperm. Specific protamine combination may form a “protamine code’’, just similar to the ‘histone code”. This suggested a potential role for PTMs on sperm histones and protamine sin epigenetic signatures underlying transgenerational inheritance [119 Brunner 2014].
Roles of spermatozoal RNAs
Spermatozoa contain many specific RNAs, mRNAs, miRNA spiwi interacting RNA’s (piRNAs), which could be useful for embryo development by modifying gene expression upon fertilization [120,121]. With use of reverse transcriptase polymerase chain reaction, in situ hybridization, along with oligo DNA microarray hybridization have allowed the identification of certain mRNAs in mature human spermatozoa some of which encode protamine and hormonal receptors [122]. Spermatozoa RNAs are markers of male infertility especially concerning spermatogenesis. Abnormally increased mRNA retention in ejaculated sperm associated with protamine translation dysregulation, protamine deficiency in sperm along with infertility [123]. P1 and P2 transcript levels are also reported to be lower in ejaculated sperms of asthenozoospermic men [123] has been suggested that sperm RNAs playa dynamic role and contribute to stabilizing the nuclear envelope and the interaction between DNA histone during protamine transition. Thus they may mark DNA sequences, which stay bound to histones which is important for embryo development [124]. Microarray technologies show different mRNA’s pattern between fertile and infertile men, which were associated with pregnancy rates in ART [125-127], reviewed in Boisonnass [128].
Epigenetic Reprogramming in Embryo
Prior to fertilization there is different but distinct patterns of DNA methylation and chromatin organization in sperm and egg which governs the gene expression [129]. In sperm DNA is tightly condensed by protamines. During embryo reprogramming starts with paternal genome decondensing as their protamines get replaced by maternally derived histones [129]. Immediately following fertilization paternal pronuclear genome goes through demethylation [130-136]. In maternal genome demethylation follows, so that at 4 cell stage of the embryo the DNA methylation status of 2 paternal genomes is equalized [136]. Epigenetic marks at imprinted genes are protected by specific proteins from epigenetic reprogramming throughout the preimplantation development of embryo [43].
Commitment for lineage
Once blastocyst is formed the inner cell mass (ICM) and trophectoderm each carry distinct epigenetic signatures [129]. Decreasing levels of the 5 hydroxyl methyl cytosine (5 hmC) and TET oxidases are associated with progressive differentiation and silencing the genes responsible for early development [137,138]. Distinct epigenetic signatures would have been acquired by the time cells become fully committed to their lineage which reflects their phenotype, developmental history along with environmental influences [129]. Trophectoderm derived placental cells, remain relatively hypo methylated as compared to derivatives of ICM [139], with role of placenta in endometrial invasion along with lesser need for marked differentiation along with longevity [129].
Human Diseases Associated with Epigenetic D Dysregulation
Dysregulation of epigenetic control mechanisms leads to disruption of multiple organ systems. Mutations in proteins which establish, remove or recognize various epigenetic marks may negatively affect the development of multiple organ systems, which is seen in multiple human genetic syndromes like ATRX (α thalassemia, mental retardation) Rubinstein Taylor [OMIN180849, CREBBP/EP300] [140], CHARGE (CHD7) and Reit syndrome [OMIM 312750, MECP2] [141]. Epimutation is an aberrant DNA methylation/histone modification pattern. These alterations happen in presence or absence of underlying genomic change and thus are known as primary and secondary epimutation, respectively. Primary epimutation leads to aberrant erasure or maintenance of epigenetic marks. Non imprinted genes somatic epimutation, which occur due to mitotic abnormalities in the normal maintenance of epigenetic marks can lead to abnormal growth regulation, e.g., is 20% of breast cancer shows hyper methylation of the BRCA1 promoter, in combination with an inherited/somatic mutation on 2nd allele [142]. Decrease growth potential also results from an epimutation. Promoter methylation of wingless type MMTV integration site family member 2 (WNT2) in placenta is associated with decreased birth weight percentile in neonate, showing a single epigenetic involvement can affect female phenotype [143].
In Human Disease and Imprinting: There is more complicated epigenetic regulation for imprinted genes as compared to non-imprinted genes, giving more chance to acquired epigenetic errors. Parental contribution of specific imprinted regions access genome can get disrupted by a number of different mechanisms including genetic and/or epigenetic change. Primary epimutation can occur from errors in reprogramming in imprinting. Failure to erase imprint leads to genome of one sex which carries imprint of the other sex. On fertilization this gametes leads to a zygote carrying uniparental imprints on both the parental chromosomes. Failure to establish appropriate imprints in the sperms or oocytes may lead to various disorders of growth and development which includes infertility [90]. Uniparental disomy (UPD) is one of the genomic alterations which lead to imprinting disorders, where both homologues of a chromosome segment or a region, gets inherited from only one parent, with no contribution from opposite parent. UPD at specific areas containing imprinted regions can cause imprinted disorders whereas from nonimprinted genome are not associated with disease [144]. UPD of any chromosome is believed to occur in 1:3500 live births [145]. Deletion or duplication of imprinted genomic area can change the parental contribution that is 2nd way how these imprinted genes may be regulated. There are imprinted genes get expressed in placenta, and have functional role in fetal growth retardation and brain development [146].
Thus epigenetic and associated genetic changes at imprinted genes lead to human disease which often shows aberrant growth and an abnormal neurodevelopment. Neurologic and psychiatric disorders in a deregulation of imprinted genes are involved with effects if parents of origin ,one set involved added in PWS Angelmann syndrome(AS)two very gene 15q-t P11.q13 [86]. Paternal deletion of C15q11-13 leading to PWS, while a maternal deletion of C 15q11-13 leading to AS [39].
One of the pediatric growth disorders because of abnormal imprinting is Beckwith-Wiedman syndrome (BWS). BWS Patients present with macroglossia, macrosomia, visceromegaly; embryonal tumors like hepatoblastoma, neuroblastoma, Wilms tumor, rhabdomyosarcoma, omphalocoele, neonatal hypoglycemia, ear creases, pits, adrenocortical cytomegaly and renal abnormalities [35]. Pregnancies where fetus has BWS, are usually associated with polyhydramnios, huge placenta, very long thickened umbilical cord and a risk of preterm delivery [148,149]. This is also associated with mesenchyme dysplasia of placenta [88]. There are 2 different epimutation hypo methylation of the proximal IC on the maternal chromosome along with hyper methylation of the distal IC on the maternal chromosome. Hyper methylation at IC2 can occurs as a secondary epimutation if there is an underlying deletion of maternal IC2 region.
When BWS occur with paternal UPD of C 11P15.5 genomic imprints at both in 1C1 and IC2 get affected [41]. There are epigenome types–phenotype correlations exhibited by BWS patients e.g., tumor risk is seen with patients with BWS, with molecular abnormality, which include down regulation of IC1.
Russel Silver syndrome (RSS) is a syndrome where both pre and post natal growth are affected, patients having dysmorphic features, along with different presence of limb: body asymmetry along with developmental delay. 2 different epigenetic defects have been seen in RSS till date [150] 1st is maternal UPD of C 7, which is present in 10% of RSS cases [151]. Hypo methylation at IC1 on the paternal C 11p15.5 is seen in 45% of patients [150-154]. This is opposite both in phenotype as well as genotype to BWS with gain in methylation occurring in same region. Funnily some males with oligo zoospermia are seen to lack parental methylation in IGF2/H191C1 locus on Cp11p15.5, in their sperm [155]. Thus children inheriting the C from father are at greater risk of developing the risk of developing RSS [150].
Imprinting disorders include imprinting errors at multiple imprinted domains e.g., in both BWS and RSS, some patients exhibited loss of methylation, not only at IC1on Cp11p15.5-IC1 for RSS and IC2for BWS but also show this at other loci. Why multilocus loss of methylation (MLOM), occurs is unclear. This also occurs is transient neonatal diabetes mellitus, having initial defect in PLAGL1DMR on C6q24, which is associated with LOM at other DMRs [156,157]. It is believed that this MLOM is due to homozygous mutation in the ZFP57 gene. ICRs are genetically divergent across species despite genomic imprinting being conserved in mammals. Hur et al. to study if ICRs play a species specific role in regulating imprinting at a given locus examined the H19/Igf2 imprinted locus, the misregulation of which is associated with BWS and SRS. They generated a knockout in which the endogenous H19/Igf2 ICR (mICI) is replaced by the orthologous human ICR (hICI) sequence called H19 (hICI). They showed that hICI concentration functionally replaces mICI on the maternal allele. In contrast paternally transmitted h1CI lead to growth restriction, abnormal hICI methylation and loss of H19 and IGF2 imprinted expression. Imprint establishment at hi C1 is impaired in the male germ line which is associated with an abnormal composition of histone post translational modification as compared to m1CI. Thus this study revealed evolutionary divergent paternal imprinting at ICI between mice and humans. Conserved maternal imprinting mechanism and function of ICI shows the possibility of modeling maternal transmission of hiCI mutations associated with BWS in mice. They further proposed the analysis in paternal knockout H19 (+hiCI) mice to elucidate molecular mechanisms which may underlie SRS [158]. This gene encodes an oocyte derived maternal factor which takes part in preimplantation maintainance of imprints at multiple loci.
Mutations in the NLRP gene are also associated with imprinting errors helping at multiple genomic loci. Being members of CATERPILLER family of proteins, NLRP proteins are involved in inflammation and apoptosis [39]. Mutations of NLRP7 in the oocyte are associated with aberrant imprinting at multiple loci, which causes formation of partial hydatidiform mole where the term ‘biparental’ is used because of origin from both oocyte and sperm.
Art in Epigenetics: Increasing incidence of BWS [159,161-165] and AS [160] is observed in some studies in children conceived with ART. Some studies contradict that [165]. Further Vermeiden et al. reviewed if these imprinting disorders got increased following IVF or ICSI [166]. There can be problem at level of ovulation inductions [167]. Fauque et al. at the level of in vitro culture of gametes and embryos [168]. Umdhane et al. to study the hypothesis regarding after ICSI some embryos fail to develop normally and they may have imprinting disorders which are associated with developmental failure they analyzed the methylation profile of H19DMR, a paternally imprinting control region in high grade blastocyst, in couples where they are available. They found marked hypomethylation in the paternal allele in half of the embryos, independently of the stage which they were arrested (morula, compacted morula, preblastocysts or graded blastoctsts). Conversely same embryos showed significant methylation on the maternal allele whereas few others showed both hypomethylation of the paternal allele and abnormal methylation of the maternal allele. The matching sperm at the origin of the embryo exhibited normal methylation patterns. Thus, hypomethylation of the paternal alleles in the embryo does not seem to be inherited in the early embryo. Analysis of a few oocytes suggests that the defects in erasure of the allele in some embryos. None of these imprinting alterations could be related to a particular stage of developmental arrest, compared with the high grade blastocysts, embryos with development failure are more likely to have an abnormal imprinting at H19 (p < 0.01) [169].
In gametes a methylation at imprinting control regions (ICRs) gets established in a sex specific manner, and needs to be maintained stably during development in somatic cells to ensure the correct mammalian expression of imprinted genes. Besides DNA methylation, these ICRs are marked by specific histone modifications. If these marks are essential for maintainance of genome imprinting is unclear. Zhang et al. showed that histone H3 lysine 9methylases H9αand GTP are required for stable maintainance of imprinted DNA methylationin embryonic stem cells. However their catalytic activity and the G9α/GLP dependent H3K9me2 mark are completely dispensable for imprinting maintenance, despite the genome wide loss of non imprinte DNA methylation in H3K9me2 depleted cell. They provided additional evidence that the G9α/GLP complex protects imprinted DNA methylation by recruitment of DNA methyl transferases, which antagonize TET deoxygenase-dependent erosion of DNA methylation at ICRs [170].
Conclusions
Basically epigenetic mechanisms control development and regulate gene expression in various types of cells of the organism, each carrying similar DNA sequence. In simple language, nucleotide of DNA are letters of the complicated text and these epigenetic labels or marks are the spaces, punctuations, sentences, paragraphs and styles which gives meaning to this complicated text. Here we have tried to discuss the epigenetic markers like DNA methylation at CpG dinucletotide, covalent modifications of histone proteins and details of noncoding RNAs including long ncRNAs, miRNA. Besides importance of lncRNAs has been discussed in dosage compensation in X linked genes with role of Xi and further the value of genome imprinting in ART and various disorders like BWS, RSS or both. Further role of environmental stressors in stresses. Role of famines in both Chinese as well as Dutch famines highlights immediate changes [3,6,156] and effects on future generations [171,172]. Epigenetic information can be inherited through the mammalian germ line and represents a plausible Tran’s generational carrier of environmental information. Carone et al. carried out expression profiling screen for genes in mice that responded to paternal diet. Offspring of males fed a low protein diet exhibited increased hepatic expression of many genes involved in lipid and cholesterol biosynthesis and decreased levels of cholesterol esters, relative to the offspring of males fed a control diet. Epigenomic profiling of offspring livers revealed numerous modest 20% conjugates in cysteine methylation, depending on paternal diet including reproductive changes in methylation over a liker or enhancer for the key lipid regulator para. These results in conjunction with recent epidemiological data indicate that paternal diet can affect cholesterol and lipid metabolism in offspring and define a model to study environment reprogramming in the human genome [173]. Environmental pollutants and medications may affect fetal epigenetic marks e.g., is in choline intake in pregnancy increased placental promoter methylation of cortisol regulation genes CRH and NR3I leading to improved stress response in children by lowering cortisol levels in H-P-A axis. Thus future of epigenetics research lies in understanding the effects of interaction of epigenetics and environment emphasis on fetal programming, understanding and uncovering role of medicine and nutrition and assesses risk for adult onset disease.
References
- Waterland RA, Michels KB (2007) Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 27: 363-388.
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, et al. (2011) Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471: 68-73.
- Huang C, Li Z, Wang M, Martorell R (2010) Early life exposure to the 1959-1961 Chinese famine has long-term health consequences. J Nutr 140: 1874-1878.
- Kaur KK, Allahbadia GN, Singh M (2016) Short Commentary-‘On induced pluripotent stem cells with special empasis on trnscriptomics and recent advances in therapeutic potential”. Transcriptomics.
- Mathers JC (2007) Early nutrition: impact on epigenetics. Forum Nutr 60: 42-48.
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, et al. (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 105: 17046-17049.
- Chawla RK, Watson WH, Jones DP (1996) Effect of hypoxia on hepatic DNA methylation and tRNA methyltransferase in rat: similarities to effects of methyl-deficient diets. J Cell Biochem 61: 72-80.
- Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, et al. (2008) Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun 372: 892-897.
- Yang J, Ledaki I, Turley H, Gatter KC, Montero JC, et al. (2009) Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann N Y Acad Sci 1177: 185-197.
- Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, et al. (2009) Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep 10: 1235-1241.
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, et al. (2007) Distribution,silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457-466.
- ENCODE Project Consortium (2004) The ENCODE (Encyclopaedia Of DNA Elements) Project. Science 306: 636-640.
- Maunakea AK, Chepelev I, Zhao K (2010) Epigenome mapping in normal and disease States. Circ Res 107: 327-339.
- Lande-Diner L, Zhang J, Ben-Porath I, Amariglio N, Keshet I, et al. (2007) Role of DNA methylation in stable gene repression. J Biol Chem 282: 12194-12200
- ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57-74.
- Cedar H, Bergman Y (2012) Programming of DNA methylation patterns. Annu Rev Biochem 81: 97-117.
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, et al. (2012) Landscape of transcription in human cells. Nature 489: 101-108.
- Kaikkonen MU, Lam MT, Glass CK (2011) Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res 90: 430-440.
- Grewal SI (2010) RNAi-dependent formation of heterochromatin and its diverse functions. Curr Opin Genet Dev 20: 134-141.
- Chuang JC, Jones PA (2007) Epigenetics and microRNAs. Pediatr Res 61: 24R-29R.
- Carthew RW, Sontheimer EJ (2009) Origins and Mechanisms of miRNAs and siRNAs. Cell 136: 642-655.
- Kaur KK, Allahbadia GN, Singh M (2016) An Update on MicroRnas? And metabolic regulation with Future Therapeutic Potentials Regarding Diagnosis of Obesity, Metabolic Syndrome & Other Related Disorders. Metabolonomics.
- Sheik Mohamed J, Gaughwin PM, Lim B, Robson P, Lipovich L (2010) Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA 16: 324-337.
- Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136: 629-641.
- Moran VA, Perera RJ, Khalil AM (2012) Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res 40: 6391-6400.
- Lambeth LS, Raymond CS, Roeszler KN, Kuroiwa A, Nakata N, et al (2014) Overexpression of DMRT1induces the male pathway in embryonic chicken gonad. Developmental Biology 389:160-172.
- RoeszlerN, Itman C, Sinclair AH, Smith CA (2012) The long noncoding RNA MHM plays a role in chicken embryonic development including gonadogenesis. Dev Biol 366:317-326.
- Rastetter RH, Smith CA, Wilhelm D (2015) The role of non-coding RNAs in male sex determination and differentiation. Reproduction 150: R93-107.
- Heard E, Deutsche CM (2006) Dosage compensation in mammals: fine tuning the expression of the X Chromosome. Genes Dev 20: 1848-67.
- Pessia E, Makino T, Bailly-Bechet M, McLysaght A, Marais GA (2012) Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc Natl Acad Sci USA 109: 5346-5351.
- Morey C, Avner P (2010) Genetics and epigenetics of the X chromosome. Ann N Y Acad Sci 1214: 18-33.
- Okamoto I, Heard E (2009) Lessons from comparative analysis of X-chromosome inactivation in mammals. Chromosome Res 17: 659-669
- Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. Nat Rev Genet 2: 21-32.
- Mutter GL (1997) Role of imprinting in abnormal human development. Mutat Res 396: 141-147.
- Choufani S, Shuman C, Weksberg R (2010) Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 154: 343-354.
- Tycko 1, Morison IM (2002) Physiological functions of imprinted genes. J Cell Physiol 192: 245-258.
- Reik W, Constância M, Fowden A, Anderson N, Dean W, et al. (2003) Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol 547: 35-44.
- Weksberg R (2010) Imprinted genes and human disease. Am J Med Genet C Semin Med Genet 154C: 317-320.
- Horsthemke B (2010) Mechanisms of imprint dysregulation. Am J Med Genet C Semin Med Genet 154C: 321-328.
- Choufani S,Shapiro JS, Susiarjo M, Butcher DT, Lou Y, et al. (2011) A novel approach identifies new differentially methylated regions (DMR’s) associated with imprinted genes. Genome Res 21:465-76.
- Gabory A, Jammes H, Dandolo L (2010) The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays 32: 473-480.
- McLaren A (2003) Primordial germ cells in the mouse. Dev Biol 262: 1-15.
- Feng S, Jacobsen SE, Reik W (2010) Epigenetic reprogramming in plant and animal development. Science 330: 622-627.
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, et al. (2010) Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463: 1101-1105.
- Maatouk DM, Kellam LD, Mann MR, Lei H, Li E, et al. (2006) DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development 133: 3411-3418.
- Napoles M, Nesterova T, Brockdorff N (2007) Early loss of Xist RNA expression and inactive X chromosome associated chromatin modification in developing primordial germ cells. PLoS ONE 2: e860.
- Sugimoto M, Abe K (2007) X chromosome reactivation initiates in nascent primordial germ cells in mice. PLoS Genet 3: e116.
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, et al. (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429: 900-903.
- Li X, Ito M, Zhou F, Youngson N, Zuo X, et al. (2008) A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15: 547-557.
- Kim K, Doi A, Wen B, Ng K, Zhao R, et al. (2010) Epigenetic memory in induced pluripotent stem cells. Nature 467: 285-290.
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N (2010) Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6: 407-411.
- Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, et al. (2011) Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol 29: 1117-1119.
- Zhao T, Zhang ZN, Rong Z, Xu Y (2011) Immunogenicity of induced pluripotent stem cells. Nature 474: 212-215.
- Gurdon JB, Wilmut I (2011) Nuclear transfer to eggs and oocytes. Cold Spring Harb Perspect Biol.
- Awe JP, Byrne JA (2013) Identifying candidate oocyte reprogramming factors using cross-species global transcriptional analysis. Cell Reprogram 15: 126-133.
- Xu F, Flowers S, Moran E (2012) Essential role of ARID2 protein-containing SWI/SNF complex in tissue-specific gene expression. J Biol Chem 287: 5033-5041.
- Houlard M, Berlivet S, Probst AV, Quivy JP, Héry P, et al. (2006) CAF-1 is essential for heterochromatin organization in pluripotent embryonic cells. PLoS Genet 2: e181.
- Gene Cards.org (2012) The genecard s Human Gene Data.
- Pivot-Pajot C, Caron C, Govin J, Vion A, Rousseaux S, et al. (2003) Acetylation-dependent chromatin reorganization by BRDT, a testis-specific bromodomain-containing protein. Mol Cell Biol 23: 5354-5365.
- Kim SK, Suh MR, Yoon HS, Lee JB, Oh SK, et al. (2005) Identification of developmental pluripotency associated 5 expression in human pluripotent stem cells. Stem Cells 23: 458-462.
- Liu YJ, Nakamura T, Nakano T (2012) Essential role of DPPA3 for chromatin condensation in mouse oocytogenesis. Biol Reprod 86: 40.
- Jude CD, Climer L, Xu D, Artinger E, Fisher JK, et al. (2007) Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 1: 324-337.
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, et al. (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311: 844-847.
- Becker M, Becker A, Miyara F, Han Z, Kihara M, et al. (2005) Differential in vivo binding dynamics of somatic and oocyte-specific linker histones in oocytes and during ES cell nuclear transfer. Mol Biol Cell 16: 3887-3895.
- Aoto T, Saitoh N, Sakamoto Y, Watanabe S, Nakao M (2008) Polycomb group protein associated chromatin is reproduced in post mitotic G1 phase and is required for S phase progression. J Biol Chem 283:18905-18915.
- Biermann K, Steger K (2007) Epigenetics in male germ cells. J Androl 28: 466-480.
- Illingworth RS, Bird AP (2009) CpG islands--'a rough guide'. FEBS Lett 583: 1713-1720.
- Lister R, Ecker JR (2009) Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res 19: 959-966.
- Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, et al. (2011) Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet 7: e1002389.
- Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H (2013) Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res 41: 738-745.
- Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293: 1089-1093.
- Rousseaux S, Caron C, Govin J, Lestrat C, Faure AK, et al. (2005) Establishment of male-specific epigenetic information. Gene 345: 139-153.
- Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3: 662-673.
- Santos F, Dean W (2004) Epigenetic reprogramming during early development in mammals. Reproduction 127: 643-651.
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, et al. (2002) Epigenetic reprogramming in mouse primordial germ cells. Mech Dev 117: 15-23.
- Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, et al. (2008) Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452: 877-881.
- Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM (2007) Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol 307: 368-379.
- Kerjean A, Dupont JM, Vasseur C, Le Tessier D, Cuisset L, et al. (2000) Establishment of the paternal methylation imprint of the human H19 and MEST/PEG1 genes during spermatogenesis. Hum Mol Genet 9: 2183-2187.
- Marques CJ, João Pinho M, Carvalho F, Bièche I, Barros A, et al. (2011) DNA methylation imprinting marks and DNA methyltransferase expression in human spermatogenic cell stages. Epigenetics 6: 1354-1361.
- Benchaib M, Braun V, Ressnikof D, Lornage J, Durand P, et al. (2005) Influence of global sperm DNA methylation on IVF results. Hum Reprod 20: 768-773.
- Houshdaran S, Cortessis VK, Seigmund K, Yang A, Laird PW, et al. (2007) Widespread epigenetic abnormalities suggest suggest a broad DNA methylation erasure defectin abnormal human sperm. PLoS ONE 2: e1289.
- Aston KI, Punj V, Liu L, Carrell DT (2012) Genome-wide sperm deoxyribonucleic acid methylation is altered in some men with abnormal chromatin packaging or poor in vitro fertilization embryogenesis. Fertil Steril 97: 285-292.
- Marques CJ, Carvalho F, Sousa M, Barros A (2004) Genomic imprinting in disruptive spermatogenesis. Lancet 363: 1700-1702.
- Marques CJ, Costa P, Vaz B, Carvalho F, Fernandes S, et al. (2008) Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. Mol Hum Reprod 14: 67-74.
- Kobayashi H, Sato A, Otsu E, Hiura H, Tomatsu C, et al. (2007) Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum Mol Genet 16: 2542-2551.
- Kobayashi H, Hiura H, John RM, Sato A, Otsu E, et al. (2009) DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet 17: 1582-1591.
- Boissonnas CC, Abdalaoui HE, Haelewyn V, Fauque P, Dupont JM, et al. (2010) Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur J Hum Genet 18: 73-80.
- Hammoud SS, Purwar J, Pflueger C, Cairns BR, Carrell DT (2010) Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil Steril 94: 1728-1733.
- Poplinski A, Tüttelmann F, Kanber D, Horsthemke B, Gromoll J (2010) Idiopathic male infertility is strongly associated with aberrant methylation of MEST and IGF2/H19 ICR1. Int J Androl 33: 642-649.
- Sato A, Hiura H, Okae H, Miyauchi N, Abe Y, et al. (2011) Assessing loss of imprint methylation in sperm from subfertile men using novel methylation polymerase chain reaction Luminex analysis. Fertil Steril 95: 129-134, 134.
- Navarro-Costa P, Nogueira P, Carvalho M, Leal F, Cordeiro I, et al. (2010) Incorrect DNA methylation of the DAZL promoter CpG island associates with defective human sperm. Hum Reprod 25: 2647-2654.
- Nanassy L, Carrell DT (2011) Abnormal methylation of the promoter of CREM is broadly associated with male factor infertility and poor sperm quality but is improved in sperm selected by density gradient centrifugation. Fertil Steril 95: 2310-2314.
- Nanassy L, Carrell DT (2011) Analysis of the methylation pattern of six gene promoters in sperm of men with abnormal protamination. Asian J Androl 13: 342-346.
- Chan D, Cushnie DW, Neaga OR, Lawrance AC, Rozen R, et al. (2010) Strain-specific defects in testicular development and sperm epigenetic patterns in 5,10-methylenetetrahydrofolate reductase-deficient mice Endocrinology 151: 3363-73.
- Khazamipour N, Noruzinia M, Fatehmanesh P, Keyhanee M, Pujol P (2009) MTHFR promoter hypermethylation in testicular biopsies of patients with non-obstructive azoospermia: the role of epigenetics in male infertility. Hum Reprod 24: 2361-2364.
- Wu W, Shen O, Qin Y, Niu X, Lu C, et al. (2010 ) Idiopathic male infertility is strongly associated with aberrant promoter methylation of methylenetetrahydrofolate reductase (MTHFR). PLoS ONE 5: e13884.
- Filipponi D, Feil R (2009) Perturbation of genomic imprinting in oligozoospermia. Epigenetics 4: 27-30.
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074-1080.
- Hazzouri M, Pivot-Pajot C, Faure AK, Usson Y, Pelletier R, et al. (2000) Regulated hyperacetylation of core histones during mouse spermatogenesis: involvement of histone deacetylases. Eur J Cell Biol 79: 950-960.
- Fenic I, Sonnack V, Failing K, Bergmann M, Steger K (2004) In vivo effects of histone-deacetylase inhibitor trichostatin-A on murine spermatogenesis. J Androl 25: 811-818.
- Fenic I, Hossain HM, Sonnack V, Tchatalbachev S, Thierer F, et al. (2008) In vivo application of histone deacetylase inhibitor trichostatin-a impairs murine male meiosis. J Androl 29: 172-185.
- Glaser S, Lubitz S, Loveland KL, Ohbo K, Robb L, et al. (2009) The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin 2: 5.
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, et al. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941-953.
- Lee MG, Wynder C, Cooch N, Shiekhattar R (2005) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437: 432-435.
- Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y (2007) Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 450: 119-123.
- Montellier E, Rousseaux S, Zhao Y, Khochbin S (2012) Histone crotonylation specifically marks the haploid male germ cell gene expression program: post-meiotic male-specific gene expression. Bioessays 34: 187-193.
- Sonnack V, Falling K, Bergmann M, Steger K (2002) Expression of hyperacetylated histone H4 During normal and impaired spermatogenesis. Andrologia 34: 384-90.
- Carrell DT, Liu L (2001) Altered protamine 2expression is uncommon in donors of known fertility but common common among men with poor fertilizing capacity and may reflect other abnormalities of spermiogenesis. J Androl 22: 604-10.
- Aoki VW, Emery BR, Liu L, Carrell DT (2006) Protamine levels vary between individual sperm cells of infertile human males and correlate with viability and DNA integrity. J Androl 27: 890-898.
- Aoki VW, Moskovtsev S, Willis J, Liu L, Mullen JB, et al. (2005) DNA integrity is compromised in protamine-deficient human sperm. J Androl 26: 741-748.
- Aoki VW, Liu L, Jones KP, Hatasaka HH, Gibson M, et al. (2006) Sperm protamine 1/protamine 2 ratios are related to in vitro fertilization pregnancy rates and predictive of fertilization ability. Fertil Steril 86: 1408-1415.
- Torregrosa N, Domínguez-Fandos D, Camejo MI, Shirley CR, Meistrich ML, et al. (2006) Protamine 2 precursors, protamine 1/protamine 2 ratio, DNA integrity and other sperm parameters in infertile patients. Hum Reprod 21: 2084-2089.
- Carrell DT, Emery BR, Hammoud S (2007) Altered protamine expression and diminished spermatogenesis: what is the link? Hum Reprod Update 13: 313-327.
- Oliva R (2006) Protamines and male infertility. Hum Reprod Update 12: 417-435.
- Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, et al. (2009) Distinctive chromatin in human sperm packages genes for embryo development. Nature 460: 473-8.
- Carrell DT, Hammoud SS (2009) The human sperm epigenome and its potential role in embryonic development. Mol Hum Reprod 16: 37-47.
- Chevaillier P, Mauro N, Feneux D, Jouannet P, David G (1987) Anomalous protein complement of sperm nuclei in some infertile men. Lancet 2: 806-7.
- Brunner AM, Nanni P, Mansuy IM (2014) Epigenetic marking of sperm by post-translational modification of histones and protamines. Epigenetics Chromatin 7: 2.
- Ostermeier GC, Dix DJ, Miller D, Khatri P, Krawetz SA (2002) Spermatozoal RNA profiles of normal fertile men. Lancet 360: 772-777.
- Hamatani T (2012) Human spermatozoal RNAs. Fertil Steril 97: 275-281.
- Dadoune JP (2009) Spermatozoal RNAs: what about their functions? Microsc Res Tech 72: 536-551.
- Aoki VW, Liu L, Carrell DT (2006) A novel mechanism of protamine expression deregulation highlighted by abnormal protamine transcript retention in infertile human males with sperm protamine deficiency. Mol Hum Reprod 12: 41-50.
- Kempisty B, Depa-Martynow M, Lianeri M, Jedrzejczak P, Darul-Wasowicz A, et al. (2007) Evaluation of protamines 1 and 2 transcript contents in spermatozoa from asthenozoospermic men. Folia Histochem Cytobiol 45 Suppl 1: S109-113.
- Lalancette C, Miller D, Li Y, Krawetz SA (2008) Paternal contributions: new functional insights for spermatozoal RNA. J Cell Biochem 104: 1570-1579.
- Garrido N, Martinez –Conejero JA, Jauregui J, Horcajadas JA, Simon C, et al. (2009) Microarray analysis in sperm from fertile and infertile men without basic sperm analysis abnormalities reveals a significantly different transcriptome. Fertil Steril 91: 1307-10.
- Montjean D, De La Grange P, Gentien D, Rapinat A, Belloc S, et al. (2012) Sperm transcriptome profiling in oligozoospermia. J Assist Reprod Genet 29: 3-10.
- Boissonnas CC1, Jouannet P, Jammes H (2013) Epigenetic disorders and male subfertility. Fertil Steril 99: 624-631.
- Morgan HD, Santos F, Green K, Dean W, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Gene 14: 47-58.
- Santos F, Hendrich B, Reik W, Dean W (2002) Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol 241: 172-182.
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T (2000) Demethylation of the zygotic paternal genome. Nature 403: 501-502.
- Dean W, Santos F, Stojkovic M, Zakhartchenko V, Walter J, et al. (2001) Conservation of methylation reprogramming in mammalian development:aberrant reprogramming in cloned embryos. Proc Natl Acad Sci USA 98: 13734-8.
- Beaujean N, Hartshorne G, Cavilla J, Taylor J, Gardner J, et al. (2004) Non-conservation of mammalian preimplantation methylation dynamics. Curr Biol 14: 266-267.
- Fulka H, Mrazek M, Tepla O, Fulka J Jr (2004) DNA methylation pattern in human zygotes and developing embryos. Reproduction 128: 703-708.
- Lepikhov K, Wossidlo M, Arand J, Walter J (2010) DNA methylation reprogramming and DNA repair in the mouse zygote. Int J Dev Biol 54: 1565-1574.
- Hales BF, Grenier L, Lalancette C, Robaire B (2011) Epigenetic reprogramming from gametes to blastocyst. Birth Defects Res A Clin Mol Teratol 191: 652-65.
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, et al. (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473: 398-402.
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, et al. (2011) Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8: 200-213.
- Chapman V, Forrester L, Sanford J, Hastie N, Rossant J (1984) Cell lineage-specific under methylation of mouse repetitive DNA. Nature 307: 284-286.
- Munshi A, Shafi G, Aliya N, Jyothy N (2009) Histone modifications dictate specific biological readouts. J Genet Genomics 154: 335-42.
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, et al. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185-188.
- Hansmann T, Pliusch G, Leubner M, Krol P, Endt D, et al. (2012) Constitutive promoter methylation of BRCA1and RADS1C in patients with familial ovarian cancer and early onset sporadic breast cancer. Hum Mol Genet 21: 4669-79.
- Ferreira JC, Choufani S, Grafodatskaya D, Butcher DT, Zhao C, et al. (2011) WNT2 promoter methylation in human placenta is associated with low birthweight percentile in the neonate. Epigenetics 6: 440-449.
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ (2012) Prader-Willi syndrome. Genet Med 14: 10-26.
- Yamazawa K, Ogata T, Ferguson-Smith AC (2010) Uniparental disomy and human disease: an overview. Am J Med Genet C Semin Med Genet 154: 329-334.
- Robinson WP (2000) Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 22: 452-459.
- Buiting K (2010) Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 154C: 365-376.
- Weng EY, Moeschler JB, Graham JM Jr (1995) Longitudinal observations on 15 children with Wiedemann-Beckwith syndrome. Am J Med Genet 56: 366-373.
- Wilson M, Peters G, Bennetts B, McGillivray G, Wu ZH, et al. (2008) The clinical phenotype of mosaicism for genome-wide paternal uniparental disomy: two new reports. Am J Med Genet A 146: 137-148.
- Eggermann T (2010) Russell-Silver syndrome. Am J Med Genet C Semin Med Genet 154C: 355-364
- Monk D, Bentley L, Hitchins M, Myler RA, Clayton-Smith J, et al. (2002) Chromosome 7p disruptions in Silver Russell syndrome: delineating an imprinted candidate gene region. Hum Genet 111: 376-387.
- Horike S, Ferreira JC, Meguro –Honike M, Choufani S, Smith AC, et al. (2009) Screening of DNA methylation at the H19 promoter of the distal region of its ICR1 ensures efficient detection of chromosome 11p15epimutationsin Russell-Silver syndrome. Am J Med 149: 2415-2.
- Netchine L, Rossinol S, Dufoug MN, Azzi S, Rousseau A, etal. (2007)11p15imprinting centre region 1 loss of methylation is a common and specific cause of typical Russell-silver syndrome:clinical scoring system and epigenetic phenotype correlations. J Clin Endocrinol Metab 92: 3148-54.
- Bartholdi D, Krajewsa-Walasek M, Ounap K, Gapar H (2009) Epigentic mutations in the imprinted IGF2/H19 domain I Silver Russell Syndrome: results from a large cohort of patients with SRS and SRS-like phenotype. J Med Genet46: 192-7.
- Tobi EW, Slag boorn PE, Van Dongen J, Kremer D, Stein AD, et al. (2012) Prenatal famine and genetic variation are independently and additively associated with DNA methylation at regular loci within IGF2/H19. PLoS ONE 7: e37933.
- Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, et al. (2008) Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet 40: 949-51.
- Hur SK, Freschi A, Ideraabdullah F, Thorvaldsen JL, Luense LJ, et al. (2016) Humanized H19/Igf2 locus reveals diverged imprinting mechanism between mouse and human and reflects Silver-Russell syndrome phenotypes. Proc Natl Acad Sci U S A 113: 10938-10943.
- F, Riccio A, Bartolomei MS (2016) HumanizedH19/Igf2locus reveals diverged imprinting mechanisms between mouse and human and reflects Silver Russell syndrome phenotypes. Proc Natl Acad Sci USA 113: 10938-43.
- Mackay DJ, Temple IK (2010) Transient neonatal diabetes mellitus type 1. Am J Med Genet C Semin Med Genet 154C: 335-342.
- Hiura H, Okae H, Miyauchi N, Sato F, Sato A, et al. (2012) Characterization of DNA methylation errors in patients with imprinting disorders conceived by assisted reproduction technologies. Hum Reprod 27: 2541-2548.
- Eroglu A, Layman LC (2012) Role of ART in imprinting disorders. Semin Reprod Med 30: 92-104.
- Sutcliffe AG, Peters CJ, Bowdin S, Temple K, Reardon W, et al. (2006) Assisted reproductive therapies and imprinting disorders--a preliminary British survey. Hum Reprod 21: 1009-1011.
- Chang AS, Moley KH, Wangler M, FeinbergAP, Debaun MR (2005) Association between Beckwith –Wiedman Syndrome and assisted reproductive technologies: a case series of 19 patients. Fertil Steril 83: 349-354.
- Zheng HY, Shi XY, Wu FR, Wu YQ, Wang LL, et al. (2011) Assisted reproductive technologies do not increase risk of abnormal methylation of PEG1/MEST in human early pregnancy loss. Fertil Steril 96: 84-89.
- DeBaun MR, Niemitz EL, Feinberg AP (2003) Association of In Vitro Fertilization with Beckwith-Wiedemann Syndrome and Epigenetic Alterations of LIT1 and H19. Am J Hum Genet 72: 156-60.
- Metwally M, Ledger WL (2011) Long-term complications of assisted reproductive technologies. Hum Fertil (Camb) 14: 77-87. Vermeiden JP, Bernardus RE (2013) Are imprinting disorders more prevalent after human in vitro fertilization or intracytoplasmic sperm injection? Fertil Steril 99: 642-651.
- Fauque P (2013) Ovulation induction and epigenetic anomalies. Fertil Steril 99: 616-623.
- El Hajj N, Haaf T (2013) Epigenetic disturbances in in vitro cultured gametes and embryos: implications for human assisted reproduction. Fertil Steril 99: 632-641.
- Ibala-Romdhane S, Al-Khtib M, Khoueiry R, Blachère T, Guérin JF, et al. (2011) Analysis of H19 methylation in control and abnormal human embryos, sperm and oocytes. Eur J Hum Genet 19: 1138-1143.
- Zhang T, Termanis A, Özkan B, Bao XX, Culley J, et al. (2016) G9a/GLP Complex Maintains Imprinted DNA Methylation in Embryonic Stem Cells. Cell Rep 15: 77-85.
- Li Y, He Y, Qi L, Jaddoe VW, Feskens EJ, et al. (2010) Exposure to the Chinese famine in early life and the risk of hyperglycemia and type 2 diabetes in adulthood. Diabetes 59: 2400-2406.
- Stein AD, Kahn HS, Rundle A, Zybert PA, van der Pal-de Bruin K, et al. (2007) Anthropometric measures in middle age after exposure to famine during gestation: evidence from the Dutch famine. Am J Clin Nutr 85: 869-876.
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, et al. (2010) Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143: 1084-1096.
- Jiang X, Yan J, West AA, Perry CA, Malyshevia OV, et al. (2012) Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J 26: 3563-74.

