Keywords
BRAT1; Lethal neonatal rigidity; Oncogene; Progressive encephalopathy; Sequencing
Introduction
BRAT1 (BRCA1-associated ATM activator-1) gene was initially cloned by Aglipay et al. in 2006, which they called BAAT1 [1]. This ubiquitously expressed gene encodes a protein that interacts with the BRCA1 (breast cancer 1) and ATM (ataxia telangiectasia mutated) proteins. It is involved in DNA damage response, mitochondrial function, cell proliferation, and is necessary for protein stability of PIKKs.
As other oncogenes, the BRAT1 gene has been recently related to neuronal growth. Hartz mapped the BRAT1 gene to chromosome 7p22.3 in 2012 [2], and in the same year, the involvement of this gene in brain development was confirmed by exome sequencing in patients with lethal and progressive epileptic encephalopathy [3]. This syndrome is characterized by a progressive encephalopathy with intractable seizures, rigidity, progressive microcephaly, dysautonomia, and early lethality.
Next, we will describe the known BRAT1 functions, its role in brain development, the result of BRAT1 losses in childhood, and the advances supported by NGS in the knowledge of oncogene functions and neurodevelopmental disorders.
BRAT1 Functions
BRAT1 was initially isolated as a BRCA1 binding protein, interacting with the BRCT domain of BRCA1, a well-known oncogene related to tumorigenesis and DNA damage response (DDR) [1]. This BRAT1/ BRCA1 interaction is necessary for BRCA1´s functions. Other studies have shown that BRAT1 also interacts with ATM and DNAPKs, implicated in DNA repair and DDR in general [1,4,5]. BRAT1 is required for ATM Ser phosphorylation; indeed, phosphorylation of ATM, cardinal for activation of is catalytic function induced by DNA damage, is decreased in BRAT1 knockdown cells. The ATM protein is a member of PIKKs involved in DNA repair, cell growth, and neural stem cell differentiation. Numerous different mutations in the ATM gene have been identified in patients with immunodeficiency, leukemia, lymphoma or ataxia-telangiectasia [6,7].
BRAT1 is also involved in cell growth and apoptosis [1]. Apoptotic activity is increased BRAT1 knockdown mouse embryogenic fibroblasts and human osteosarcoma cells. BRAT1 is required for cellular proliferation; the loss of BRAT1 expression significantly reduces cell proliferation and migration in BRAT1 knockdown cancer cell lines. Akt/Erk activity regulates a wide variety of cellular processes like cell proliferation, differentiation, survival and cell transformation of tumor cells. Akt/Erk´s phosphorylation status and function are decreased in BRAT1 knockdown cancer cell lines, suggesting the important function of BRAT1 in these cell processes.
In addition, BRAT1 plays an important role in cellular metabolism, particularly in regulating mitochondrial functions [8]. Glucose consumption, high levels of mitochondrial reactive oxygen species, worse mitochondrial membrane potential, reduced pyruvate dehydrogenase activity, and diminished production of ATP from mitochondria in BRAT1 knockdown cancer cells suggest it. These roles of BRAT1 in cell growth and metabolism could explain the clinical features of patients with homozygous or compound heterozygous BRAT1 mutations.
BRAT1 is required for protein stability of PIKKs, such as ATM, DNA-PK, mTOR, and mTOR-related proteins [9]. BRAT1 binds to mTORC1 complex and is required for protein stability and regulation of mTOR signaling. The mammalian target of the rapamycin (mTOR) pathway plays central roles in synaptic protein synthesis, and its dysregulation is linked to cancer, epilepsy, psychiatric disorders, and neurodevelopmental disorders [10,11]. The mTOR pathway is a central regulator of cell growth, proliferation, survival, and cap-dependent protein translation. In the brain, it plays a cardinal function in dendritic spine development and synaptogenesis [12]. mTOR complex regulates neuronal protein synthesis and actin cytoskeleton. It is involved in different cellular processes like cellular metabolism, oxidative stress, autophagy, cell proliferation, differentiation, and migration. In the brain, the dysfunction of the mTOR pathway is supposed to be associated with atypical neuronal morphology, dysfunctional autophagy, defective connectivity, cell death, mitochondrial stress, and abnormal metabolism. mTOR complex is regulated upstream by several proteins (TSC1, TSC2, PTEN…) and controls those processes through its downstream effectors (Rho GTPases, 4E-BPs, S6K1 and 2…).
Brain and BRAT1
Numerous cases of intellectual disability, autism and/or dysmorphic features, with deletions or duplications including BRATl gene, have been described in international databases. However, all these cases have wider genetic losses or gains encompassing other genes. This circumstance and the presence of different CNVs in BRATl in normal population suggest the low haploinsufficiency of this gene. Consequently, the direct and indirect functions of BRATl on cell growth, neural cell differentiation and migration, dendritic morphology, synaptogenesis, and mitochondrial metabolism could explain the important progressive characteristic of clinical features in patients with homozygous or compound heterozygous BRATl mutations [3,13-18].
In the last years, homozygous or compound heterozygous BRAT1 mutations have been described as a new cause of severe progressive encephalopathy with neonatal onset and high patient fatality [3,13-18] (Table 1). The pathogenicity of BRAT1 homozygous mutations has been related to the lethal neonatal rigidity and multifocal seizure syndrome (MIM# 614498).
| |
Puffenberg et al.
N=2 |
Saunders et al.
N=1 |
Saitsu et al.
N=2 |
Srivastava et al.
N=1 |
Straussberg et al.
N=2 |
Mundy et al.
N=1 |
Van de Pol et al.
N=3 |
Fernández-Jaén et al.
N=1 |
| Clinical Features |
|
|
|
|
|
|
|
|
| Dysmorphic features |
- |
+ |
+ |
|
- |
+ |
+ |
- |
| Microcephaly |
+ (at birth) |
+ (at birth) |
+ (postnatal) |
|
+ (postnatal) |
+ |
+ (postnatal) |
+ (postnatal) |
| Rigidity/Hypertonia |
+ |
+ |
+ |
|
+ |
+ |
+ |
+ |
| Seizures |
+ |
+ |
+ |
|
+ |
+ |
+ |
- |
| Dysautonomia (apnea, bradycardia) |
+ |
+ |
- |
|
+ |
+ |
+ |
- |
| Brain MRI |
|
|
|
|
|
|
|
|
| Cerebral atrophy |
+ (mild hypoplasia of frontal lobes) |
- |
+ |
|
- |
+ |
+ (2 cases) |
- |
| Cerebellar atrophy |
- |
- |
+ |
|
- |
+ |
+ (2 cases) |
+ |
| Died |
Before age 4 months |
Age 5 months |
Ages 1 year and 9 months (1st case) and 3 months (2nd case) |
Alive at 8 years |
Before age 6 months |
Alive at 6 years |
|
Alive at 52 months |
| BRAT1 mutation |
Homozygous
c.638_639insA |
Homozygous
c.453_454insATCTTCTC |
Compound heterozygous
c.176T>C and c.962_963del |
Compound heterozygous
c.638_639insA and c.803+1G>C |
Homozygous
c.1173delG |
Compound heterozygous
c.294dupA and c.1925C>A |
Homozygous
c.638dup |
Compound heterozygous
c.1564G>A and c.638dup |
| Postmortem examination |
Neuronal loss and gliosis |
|
1 case:
neuron depletion and gliosis in the white matter |
|
|
|
1 case:
neuron loss and atrophy of the white matter |
|
Table 1: Clinical features, brain MRI findings and BRAT1 mutations.
Puffenberger et al. [3] described two unrelated Amish sibships with prenatal microcephaly, mild hypoplasia of the frontal lobes, multifocal seizures, hypertonia, apnea, and bradycardia [3]; postmortem examination revealed a marked neuronal loss and gliosis in frontal, occipital and temporal cortex, a reduced anterior hippocampus with neuronal loss and gliosis in CA-1 zone, and scarcity of neurons and abundant Alzheimer Type 2 astrocytes in the putamen. In the same year, Saunders et al. described a new case from Mexico with similar clinical features, including microcephaly at birth [16]; two MRI scans within the first three weeks of life were normal. This lethal neonatal rigidity and multifocal seizure syndrome was later described in Japanese siblings, born of unrelated parents, with compound heterozygous mutations in BRAT1; these authors described a postnatal microcephaly, secondary to cerebral and cerebellar atrophy in both cases [15]. The postmortem autopsy of one of them revealed a remarkable loss of neurons in the cortex and cerebellum, and moderate gliosis in the frontal lobe. Two years later, Srivastava et al. communicate another case with a compound heterozygous BRAT1 mutations in a patient with an intellectual disability that was alive at the age of 8 years; no more clinical features were included [19]. Straussberg et al. [17] described two siblings born to consanguineous Arab-Muslim parents who have mutations the BRAT1 gene, with hypertonia, seizures, progressive mild microcephaly, hypertonia, and dysautonomia [17]; however, although these two cases showed a postnatal microcephaly, brain MRI was normal in both of them. In the last months, five new cases have been reported. Mundy et al. described a patient with seizures, hypertonia, apneic episodes, and arrested head growth who remains alive at 6 years of age; brain MRI showed decreased myelination and thin corpus callosum at 3 months, and right temporal lobe encephalomalacia and cerebellar and vermis hypoplasia at 3 years [14]. Van de Pol et al. described three siblings, born to consanguineous parents, the lethal neonatal rigidity and multifocal seizure syndrome [18]; brain MRI was normal at the age of 2 months and showed severe generalized atrophy ten months later in one case; two MRI scans at the age of 2 and 3 months revealed mild hypoplasia of cerebellum and brainstem, and severe brain atrophy respectively in another one; postmortem examination demonstrated a moderate neuronal loss and strong reactive gliosis in frontal cortex, reactive astrocytes, numerous Alzheimer type 2 astrocytes in underlying white matter, and an intense loss of pyramidal neurons and gliosis in the CA1 area of the hippocampus. We identified a new case, without early lethality and seizures, but with hypertonia, severe psychomotor retardation, and postnatal microcephaly (Figure 1); two timed/spaced out brain MRIs, which were performed at the ages of 19 and 48 months, showed moderate progressive cerebellar atrophy (Figures 2 and 3) [13].
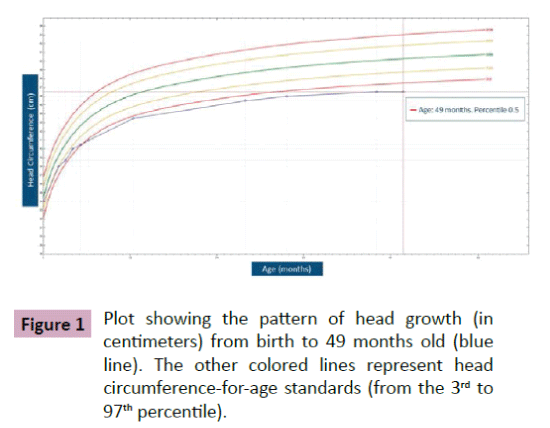
Figure 1: Plot showing the pattern of head growth (in centimeters) from birth to 49 months old (blue line). The other colored lines represent head circumference-for-age standards (from the 3rd to 97th percentile).
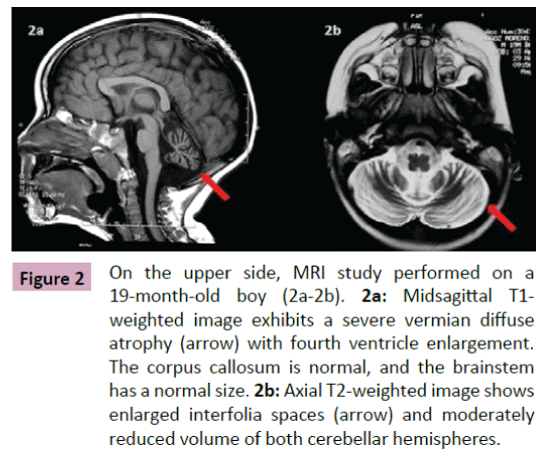
Figure 2: On the upper side, MRI study performed on a 19-month-old boy (2a-2b). 2a:Midsagittal T1-weighted image exhibits a severe vermian diffuse atrophy (arrow) with fourth ventricle enlargement. The corpus callosum is normal, and the brainstem has a normal size.2b: Axial T2-weighted image shows enlarged interfolia spaces (arrow) and moderately reduced volume of both cerebellar hemispheres
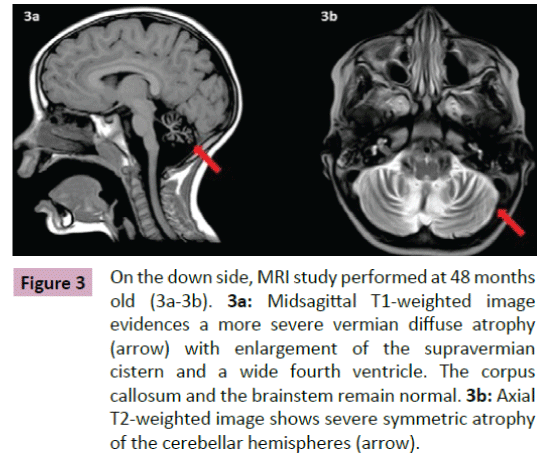
Figure 3: On the down side, MRI study performed at 48 months old (3a-3b). 3a:Midsagittal T1-weighted image evidences a more severe vermian diffuse atrophy (arrow) with enlargement of the supravermian cistern and a wide fourth ventricle. The corpus callosum and the brainstem remain normal.3b: Axial T2-weighted image shows severe symmetric atrophy of the cerebellar hemispheres (arrow).
Although other genes different to BRAT1 could contribute to the pathogenesis of the lethal neonatal rigidity and multifocal seizure syndrome, the presence of this syndrome in three pairs of siblings with confirmed homozygous BRAT1 mutations, and the rapid description of new cases with compound heterozygous or homozygous mutations in the last years support its causality. All patients suffered from seizures, hypertonia, dysautonomia and/ or psychomotor retardation. Despite to the presence of severe clinical features, cerebral MRIs only demonstrated partial or total atrophy in 8 of 12 cases, as in other progressive encephalopathies; in contrast, postmortem examination revealed marked neuron depletion and gliosis in the white matter in the 3 cases in which the autopsy was performed.
As we previously described, BRAT1 has important roles in cell proliferation processes, including cellular growth, differentiation, and tumorigenicity, and required for mitochondrial functions [8]. The BRAT1 suppression secondary to homozygous or compound heterozygous BRAT1 mutations may deteriorate cell growth and migration, influence mitochondrial homeostasis [8], and induce neuronal atrophy [3,16,18]. The presence of mild to severe arrested head growth in all cases with the neonatal rigidity and multifocal seizure syndrome and the presence of cerebral and/or cerebellar atrophy in brain MRI studies in 9 of 11 patients suggest the BRAT1 impact in cellular proliferation. The neuronal loss and gliosis of the cortex and white matter, the sparing of basal ganglia and the cells depletion of the cerebellum observed in the three cases with necropsia, confirm this hypothesis.
These features and the normal results of routine laboratory screening and extensive neurometabolic tests in the previously reported cases insinuate a more relevant role of BRAT1 in cellular proliferation and apoptosis compared with mitochondrial function. The clinical features of the lethal neonatal rigidity and multifocal seizure syndrome are shared with mitochondrial diseases [20]. Central nervous system, skeletal muscle, and heart are some of the most reliant on mitochondrial energy production and are the most symptomatic to mitochondrial defects. Intractable seizures, hypertonia, progressive encephalopathy, and dysautonomia may be observed in children with mitochondrial diseases. Besides, energetic ictal and electrical epileptogenic activity during brain development are believed to participate in the progressive cognitive deterioration or regression [21]. This suggests that the role of BRAT1 in mitochondrial function may also be important in the pathogenesis of this encephalopathic syndrome [8].
NGS, Oncogenes and Brain
NGS, including large gene panels, whole-exome sequencing (WES) and whole-genome sequencing (WGS), is rapidly advancing the precision of medical practice in diagnosis, medical management, systemic investigation and prognosis [22]. Whole exome sequencing was recently included in clinical practice and provides coverage of more than 95% of exons which contain approximately 85% of disease-causing or disease-predisposing mutations. Although NGS cannot replace a careful clinical evaluation, it is clearly changing the diagnostic algorithms.
Targeted NGS panels have been shown to increase the diagnostic yield in epilepsy, a suspected inherited ataxia, hereditary neuropathies, and myopathies [23-26]. However, in some patients, the neurological phenotype may mislead the clinician to select the wrong multi-gene panel. WES provides a potential solution to these problems. In neurodevelopmental disorders, particularly in intellectual disability and pervasive developmental disorders, diagnostic rates with WES have varied in part due to differences in the types of patients from one study to another, with rates of up to 50% [27-29].
In addition, WES has enabled the identification of previously undiagnosed diseases, novel presentations of known diseases, and specifically the discovery of new syndromes. NGS in health care has been directly involved in a rapid increase in the number of new monogenic diseases reported to OMIM. Indeed, in the last years, hundreds of new de novo and familial risk genes have been recently identified in neurodevelopmental disorders because of NGS. Dixon-Salazar et al. demonstrated the presence of twenty-two probably causative genes not previously associated with disease in the study of 118 probands with a diagnosis of a pediatric-onset neurodevelopmental disease in which most known causes had been excluded [30]. The diagnostic rate of WES in epileptic encephalopathies (EE) is also important. Veeramah et al. described the presence of de novo mutations in genes of known or plausible clinical causality in seven of ten children with refractory epilepsy and other neurological problems (autism, intellectual disability, EE) [31]; as in other neurological diseases, WES has allowed the knowledge of other causative genes in EE. Although the diagnostic yields of NGS in progressive neonatalonset encephalopathies have not been established yet, some authors have suggested that NGS will replace the current biochemical method of newborn screening or recommended the use of WGS in neonatal intensive care units, for the diagnostic approach of complex cases [16].
NGS has also been successfully applied to evaluate susceptibility, diagnosis, treatment, and prognosis of cancer [32-34]. Targeted gene panels have been more commonly used in clinical practice. Indeed, different guidelines (American Society of Clinical Oncology, National Institute for Health and Care Excellence) have recommended the use of different panels or genetic studies according to the type of cancer and the presence of family history [35]. Although the diagnostic rates of these panels range from 20 to 50%, comparable to that of WES or WGS according to some authors, WES has allowed defining previously undescribed mutations in cancer as in neurological disorders [36-38].
The advances in the knowledge of the genetic architecture that underlies neurologic and oncologic problems have shown the extensive overlap between risk genes for autism and cancer [39,40]. Chromatin remodeling and DNA repair factors (CHD family, ARID1B…), proteins involved in histone methylation (EHMT1, EHTM2, KMT2C…) or ubiquitination (UBE3A, CUL3, TBL1XR1…) have been related to cancer, neurodevelopmental disorders and even known syndromes (ARID1B in Coffin-Siris syndrome, EHMT1 in Kleefstra syndrome, UBE3A in Angelman syndrome, TBL1XR1 in Pierpont syndrome…) [41-43]. Transcription factors (FOXP family, ADNP…) are also involved in both cancer, neuronal development, autism and intellectual disability (ADNP is related with Helsmoortel-Van Der AA syndrome) [44]. Other genes involved in signal transduction pathways regulating nuclear changes (PTEN, mTOR, RAS oncogene family, AKT…) are implicated in cancer, brain development, and autism; they are the cause of known syndromes too (PTEN in Bannayan-Riley-Ruvalcaba syndrome, HRAS in Costello syndrome, AKT1 in Proteus syndrome...) [45-47].
These genetic mutations associated with cellular proliferation could affect prenatal and postnatal brain development, resulting in progressive encephalopathies, intellectual disabilities, pervasive developmental disorders or brain malformations. These aberrations could contribute to a greater susceptibility to tumors during adult life [40]. However, although some studies have demonstrated a higher tumor risk in patients with autism, other studies have paradoxically shown a decreased cancer rate in these cases [48-50].
Conclusions
NGS studies are supporting the advances in knowledge of underlying causes of tumorigenesis, normal brain development, and neurodevelopmental disorders. It is particularly interesting how some oncogenes are simultaneously involved in those processes. Genetic mutations of oncogenes linked to cell growth could modify prenatal and postnatal cerebral development, causing autism, psychomotor retardation, progressive encephalopathies or cortical dysplasias.
BRAT1 is directly or indirectly involved in cell growth, apoptosis, DNA repair, mitochondrial metabolism, and regulation of mTOR signaling (Figure 4). BRAT1 mutations are the cause of a severe progressive encephalopathy characterized by intractable epilepsy, hypertonia, arrested head growth, dysautonomia, and death. The loss of BRAT1 expression due to homozygous or compound heterozygous BRAT1 mutations leads to a deficient neuronal growth and migration, mitochondrial dysfunction, and cause neuronal atrophy. Defects in cell growth and differentiation might influence both brain development and neoplasm; some drugs targeting oncogenic pathways might also contribute to the treatment of these problems.
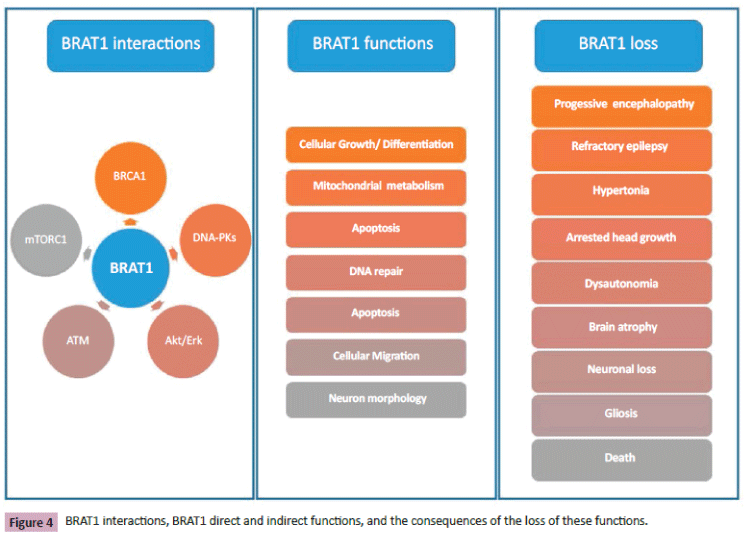
Figure 4: BRAT1 interactions, BRAT1 direct and indirect functions, and the consequences of the loss of these functions.
Acknowledgements
Part of this work was supported by R01CA79892, R01CA90631 and 5P30CA16056 from the National Institutes of Health, USA (TO).
References
- Aglipay JA, Martin SA, Tawara H, Lee SW, Ouchi T (2006) ATM activation by ionizing radiation requires BRCA1-associated BAAT1. J Biol Chem, 281: 9710-9718.
- Hartz PA (2012) Personal Communication (on 27/02/2012).
- Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, et al. (2012) Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One 7: e28936.
- Ouchi M, Ouchi T (2010) Regulation of ATM/DNA-PKcs Phosphorylation by BRCA1-Associated BAAT1. Genes Cancer 1: 1211-1214.
- So EY, Ouchi T (2011) Functional interaction of BRCA1/ATM-associated BAAT1 with the DNA-PK catalytic subunit. Exp Ther Med 2: 443-447.
- Ambrose M, Gatti RA (2013) Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood 121: 4036-4045.
- Weber AM, Ryan AJ (2015) ATM and ATR as therapeutic targets in cancer. Pharmacol Ther149: 124-138.
- So EY,Ouchi T (2014) BRAT1 deficiency causes increased glucose metabolism and mitochondrial malfunction. BMC Cancer 14: 548.
- So EY, Ouchi T (2013) The Potential Role of BRCA1-Associated ATM Activator-1 (BRAT1) in Regulation of mTOR. J Cancer Biol Res.
- Costa-Mattioli M, Monteggia LM (2013) mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci 16: 1537-1543.
- Crino PB (2015) mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb Perspect Med.
- Lipton JO, Sahin M (2014) The neurology of mTOR. Neuron 84: 275-291.
- Fernandez-Jaen A, Alvarez S, So EY, Ouchi T, Jimenez de la Pena M, et al. (2016) Mutations in BRAT1 cause autosomal recessive progressive encephalopathy: Report of a Spanish patient. Eur J Paediatr Neurol.
- Mundy SA, Krock BL, Mao R, Shen JJ (2016) BRAT1-related disease-identification of a patient without early lethality. Am J Med Genet A 170: 699-702.
- Saitsu H, Yamashita S, Tanaka Y, Tsurusaki Y, Nakashima M, et al. (2014) Compound heterozygous BRAT1 mutations cause familial Ohtahara syndrome with hypertonia and microcephaly. J Hum Genet 59: 687-690.
- Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, et al. (2012) Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med 4: 154ra135.
- Straussberg R, Ganelin-Cohen E, Goldberg-Stern H, Tzur S, Behar DM, et al. (2015) Lethal neonatal rigidity and multifocal seizure syndrome--report of another family with a BRAT1 mutation. Eur J Paediatr Neurol 19: 240-242.
- van de Pol LA, Wolf NI, van Weissenbruch MM, Stam CJ, Weiss JM, et al. (2015) Early-Onset Severe Encephalopathy with Epilepsy: The BRAT1 Gene should be Added to the List of Causes. Neuropediatrics 46: 392-400.
- Srivastava S, Cohen JS, Vernon H, Baranano K, McClellan R, et al. (2014) Clinical whole exome sequencing in child neurology practice. Ann Neurol 76: 473-483.
- Goldstein AC, Bhatia P, Vento JM (2013) Mitochondrial disease in childhood: nuclear encoded. Neurotherapeutics 10: 212-226.
- Khan S, Al Baradie R (2012) Epileptic encephalopathies: an overview. Epilepsy Res Treat, p: 403592.
- LePichon JB, Saunders CJ, Soden SE (2015) The Future of Next-Generation Sequencing in Neurology. JAMA Neurol 72: 971-972.
- Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, et al. (2015) Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 138: 276-283.
- Ream MA, Patel AD (2015) Obtaining genetic testing in pediatric epilepsy. Epilepsia 56: 1505-1514.
- Rudnik-Schoneborn S, Tolle D, Senderek J, Eggermann K, Elbracht M, et al. (2016) Diagnostic algorithms in Charcot-Marie-Tooth neuropathies: experiences from a German genetic laboratory on the basis of 1206 index patients. Clin Genet 89: 34-43.
- Todd EJ, Yau KS, Ong R, Slee J, McGillivray G, et al. (2015) Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J Rare Dis 10: 148.
- O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, et al.(2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43: 585-589.
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485: 237-241.
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, et al.(2013) Using whole-exome sequencing to identify inherited causes of autism. Neuron 77: 259-273.
- Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, et al. (2012) Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med 4: 138ra178.
- Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, et al. (2013) Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 54: 1270-1281.
- Damodaran S, Berger MF, Roychowdhury S (2015) Clinical tumor sequencing: opportunities and challenges for precision cancer medicine. Am Soc Clin Oncol Educ Book, pp: 175-182.
- Pinheiro H, Oliveira P, Oliveira C (2015) Hereditary cancer risk assessment: challenges for the next-gen sequencing era. Front Oncol 5: 62.
- Ross JS, Cronin M (2011) Whole cancer genome sequencing by next-generation methods. Am J Clin Pathol 136: 527-539.
- Dotson WD Douglas MP, Kolor K, Stewart AC, Bowen MS, et al. (2014) Prioritizing genomic applications for action by level of evidence: A horizon-scanning method. Clin Pharmacol Ther95: 394-402.
- Ley TJ, Minx PJ, Walter MJ, Ries RE, Sun H, et al. (2003) A pilot study of high-throughput, sequence-based mutational profiling of primary human acute myeloid leukemia cell genomes. Proc Natl Acad Sci USA 100: 14275-14280.
- Shen T, Pajaro-Van de Stadt SH, Yeat NC, Lin JC (2015) Clinical applications of next generation sequencing in cancer: from panels, to exomes, to genomes. Front Genet 6: 215.
- Wang L, Wheeler DA (2014) Genomic sequencing for cancer diagnosis and therapy. Annu Rev Med 65: 33-48.
- Crawley JN, Heyer WD, LaSalle JM (2016) Autism and Cancer Share Risk Genes, Pathways, and Drug Targets. Trends Genet 32: 139-146.
- Lapunzina P, Lopez RO, Rodriguez-Laguna L, Garcia-Miguel P, Martinez AR, et al. (2014) Impact of NGS in the medical sciences: Genetic syndromes with an increased risk of developing cancer as an example of the use of new technologies. Genet Mol Biol 37: 241-249.
- Heinen CA, Jongejan A, Watson PJ, Redeker B, Boelen A, et al. (2016) A specific mutation in TBL1XR1 causes Pierpont syndrome. J Med Genet.
- Nillesen WM, Yntema HG, Moscarda M, Verbeek NE, Wilson LC, et al. (2011) Characterization of a novel transcript of the EHMT1 gene reveals important diagnostic implications for Kleefstra syndrome. Hum Mutat 32: 853-859.
- Sell GL, Margolis SS (2015)From UBE3A to Angelman syndrome: a substrate perspective. Front Neurosci 9: 322.
- Krajewska-Walasek M, Jurkiewicz D, Piekutowska-Abramczuk D, Kucharczyk M, Chrzanowska KH, et al. (2016) Additional data on the clinical phenotype of Helsmoortel-Van der Aa syndrome associated with a novel truncating mutation in ADNP gene. Am J Med Genet A.
- Bhargava R, Au Yong KJ, Leonard N (2014) Bannayan-Riley-Ruvalcaba syndrome: MRI neuroimaging features in a series of 7 patients. AJNR Am J Neuroradiol 35: 402-406.
- Cohen MM (2014) Proteus syndrome review: molecular, clinical, and pathologic features. Clin Genet 85: 111-119.
- Gripp KW, Lin AE (2012) Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med 14: 285-292.
- Darbro BW, Singh R, Zimmerman MB, Mahajan VB, Bassuk AG (2016) Autism Linked to Increased Oncogene Mutations but Decreased Cancer Rate. PLoS One 11: e0149041.
- Crino PB (2011) mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 17: 734-742.
- Xue Y, Ankala A, Wilcox WR, Hegde MR (2015) Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 17: 444-451.