Xi Chen*
Center for Arrhythmia Research, University of Michigan, USA
*Corresponding Author:
Xi Chen
Center for Arrhythmia Research, University of Michigan, USA
Tel: +1 (734) 764 1817
E-mail: xichenum@umich.edu
Received date: November 10, 2016; Accepted date: November 18, 2016; Published date: November 20, 2016
Citation: Chen X. Ca2+ Homeostasis in Normal and Diseased Heart. Interv Cardiol J 2016, 2:3. doi: 10.21767/2471-8157.100036
The heart’s ability to contract greatly depends on a mechanism termed excitation-contraction coupling (E-C coupling). In each heartbeat, the open of voltage gated L-type Ca2+ channel (LTCC) due to membrane depolarization results in the influx of a small amount of Ca2+, which in turn triggers massive Ca2+ release from sarcoplasmic reticulum (SR). The binding of cytosolic Ca2+ with troponin C of myofilaments induces shortening of myofilament, so the excitatory membrane depolarization is converted into cell contraction. After contraction, 99% of Ca2+ is either recycled back to SR through SR Ca2+-ATPase (SERCA), or extruded out of the cell through Na+-Ca2+ exchanger (NCX).
Editorial
The heart’s ability to contract greatly depends on a mechanism termed excitation-contraction coupling (E-C coupling). In each heartbeat, the open of voltage gated L-type Ca2+ channel (LTCC) due to membrane depolarization results in the influx of a small amount of Ca2+, which in turn triggers massive Ca2+ release from sarcoplasmic reticulum (SR) [1]. The binding of cytosolic Ca2+ with troponin C of myofilaments induces shortening of myofilament, so the excitatory membrane depolarization is converted into cell contraction. After contraction, 99% of Ca2+ is either recycled back to SR through SR Ca2+-ATPase (SERCA), or extruded out of the cell through Na+-Ca2+ exchanger (NCX). During this process, ryanodine receptor (RyR), the Ca2+ channel inserted in SR membrane, serves as the SR Ca2+ release conduits. The communication between LTCC and RyR determines the amplitude of Ca2+ release through SR, and thus the force of contraction [2]. As Ca2+ plays a crucial role in E-C coupling and serves as a second messenger in signaling pathways, the tuning of Ca2+ release is precisely regulated by varieties of proteins, kinases, and ions. In fact, any mutation in LTCC, RyR, or other Ca2+ related protein may lead to ventricular arrhythmia, impaired contractility, or cardiomyopathy.
Catecholaminergic Polymorphism Ventricular Tachycardia (CPVT) is an inherited heart disorder, which is triggered due to cytosolic Ca2+ dysregulation. Patients have normal heart structure and function in resting state, but burst severe ventricular tachycardia morphologies when under acute emotional stress or after exercise [3]. More than 70 mutations of RyR, which are distributed in three hotspots in amino acid sequence, are known to be associated with CPVT. With gain-offunction mutations, RyRs have increased open probabilities, even when the cardiomyocyte is in resting condition during diastole. The stochastic, unsynchronized SR Ca2+ release during diastole overloads NCX to extrude Ca2+ out of the cell. The stoichiometry of 3 Na+ (in) to 1 Ca2+ (out) generates a net inward current, which depolarizes membrane potential and triggers a delayed-afterdepolarization after a normal action potential. In this situation, paroxysmal tachycardia and arrhythmia can be triggered, even the focal Ca2+ turbulence only happens in a few cardiomyocytes. While most RyR mutations lead to gain-of-function of the channel, a loss-offunction mutation (A4860G) is found in recent study, which also results in arrhythmia. Cardiomyocyte with decreased RyR activity has decreased Ca2+ transient in each beating, gradually overloading SR Ca2+. Once SR Ca2+ reaches threshold, a prolonged Ca2+ release is induced, activating NCX to trigger an early-afterdepolarization in cardiomyocyte [4].
In summary, good heart performance depends on proper Ca2+ homeostasis. Although RyR dysfunction in CPVT has been elucidated, how to rescue the phenotype remains to be investigated. As RyR acts as a scaffold for other proteins, the potential regulatory proteins, and co-factors of RyR should be studied to regulate RyR function, thus propose novel treatments for heart diseases (Figure 1).
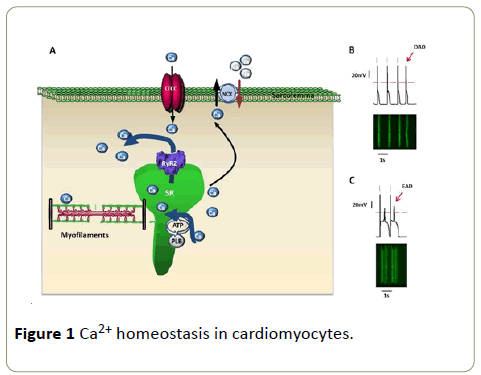
Figure 1: Ca2+ homeostasis in cardiomyocytes.
(A) Excitation-contraction coupling in cardiomyocytes. LTCC: L-type Ca2+ channel, RyR2: ryanodine receptor type 2, NCX: Na +-Ca2+ exchanger, SR: sarcoplasmic reticulum, PLB: phospholamban.
(B) Spontaneous Ca2+ release triggers delayedafterdepolarization (DAD) in cardiomyocyte. Upper panel: action potential; bottom panel: fluorescence labeled Ca2+ transient. Red bars label normal beatings of cardiomyocyte (same in figure C).
(C) Early-afterdepolarization (EAD) in cardiomyocyte.
References
- Bers DM (2002) Cardiac excitation-contraction coupling. Nature 10(415): 198-205.
- Capes EM, Loaiza R, Valdivia HH (2011) Ryanodine receptors. Skelet Muscle 1: 8.
- Loaiza R, Benkusky NA, Powers PP, Hacker T, Noujaim S, et al. (2013) Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 112: 298-308.
- Zhao YT, Valdivia CR, Gurrola GB, Powers PP, Willis BC, et al. (2015) Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc Natl Acad Sci USA 112: E1669-77.