Keywords
Antiphospholipid Syndrome Lupus Erythematosus, Systemic; Pancreatic Diseases; Pancreatitis; Pancreatitis, Acute Necrotizing
INTRODUCTION
Antiphospholipid syndrome and systemic lupus erythematosus are commonly associated. Systemic lupus erythematosus is characterized by skin manifestations such as malar rash, arthralgias, cardiac manifestations including pericarditis and endocarditis, neurologic manifestations, as well as hematological and immunological changes. Antiphospholipid syndrome is characterized by the presence of antiphospholipid antibodies with characteristic clinical manifestations. The antiphospholipid antibodies consist of lupus anticoagulant, anti-cardiolipin, and anti-beta2- microglobulin antibodies [1, 2]. The clinical manifestations of antiphospholipid syndrome include venous thromboses, arterial thromboses, thrombotic microangiopathy, and recurrent fetal loss [3]. The thromboses in antiphospholipid syndrome can affect any vascular bed, so potentially any organ system in the body can be involved. Catastrophic antiphospholipid syndrome occurs when three or more organ systems are affected by thromboses in less than a week. Catastrophic antiphospholipid syndrome occurs in only 1% of antiphospholipid syndrome patients [4]. However, it is an important clinical entity to consider due to mortality rates up to 50%.
CASE REPORT
A 20-year-old African American woman presented to the emergency center with abdominal pain, nausea, and vomiting. Her medical history is significant for systemic lupus erythematosus and antiphospholipid syndrome. Systemic lupus erythematosus was diagnosed four years prior to presentation on the basis of fever, malar rash, arthralgias, and alopecia. The patient also has hypertension and chronic kidney disease with a baseline creatinine of 3 mg/dL (reference range: 0.8-1.5 mg/dL) secondary to her underlying disease. Antiphospholipid syndrome was diagnosed one year prior to admission when she presented with a deep venous thrombosis. She has been placed on subcutaneous low-molecular weight heparin for anticoagulation for the past year, but she has not been fully compliant.
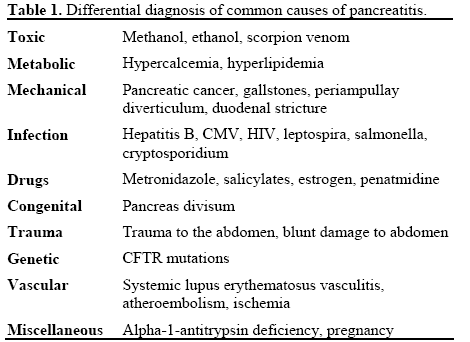
Two months prior to admission, the patient was hospitalized for acute pancreatitis. For a differential diagnosis of pancreatitis see Table 1. During her hospital course, she developed methicillin-resistant Staphylococcus aureus bacteremia, acute renal failure,and a right brachial deep venous thrombosis. Renal biopsy confirmed lupus nephritis with microangiopathic disease (Figure 1). She was discharged after re-initiation of anticoagulation with subcutaneous low-molecular weight heparin and warfarin and being apparently asymptomatic.
Figure 1. The biopsy specimen showing markedly thickened vessels,
sclerotic glomeruli, and glomeruli with Class IV glomerulonephritis.
Upon current presentation, her abdominal pain was localized to the epigastric and periumbilical regions without radiation. The pain was sharp, stabbing, and intermittent. She had vomited every other day for two weeks prior to admission, with her vomitus being nonbloody and consisting primarily of food contents. She also had diarrhea with three non-bloody, watery bowel movements the day prior to admission. Her blood pressure was elevated at 157/110 mmHg
Her laboratory data revealed lipase was 231 U/L (reference range: 6-51 U/L); amylase was 159 U/L (reference range: 30-110 U/L); anti-nuclear antibody was positive; Anti DNA titer was 1:80 (negative if less than 1:40) and equal to, or higher than, 1/2,560 SSA/Ro (positive if higher than 1/8); IgG level was normal (670 mg/dL; reference range: 751-1,560 mg/dL); C3 was 79 mg/dL (reference range: 79-152 mg/dL); C4 was 22 mg/dL (reference range: 16-38 mg/dL). Duplex ultrasound of the abdomen showed no biliary dilatation, no calculi or wall thickening in the gallbladder, negative sonographic Murphy’s sign, and no portal vein or splenic vein thrombosis. A noncontrast CT of the abdomen and pelvis after her admission was limited, but showed inflammatory changes around the pancreas with small amounts of peripancreatic fluid, thickening of the stomach wall, and free fluid in the pelvis, all consistent with pancreatitis (Figure 2). She was also found to have worsening kidney function with a creatinine of 5.3 mg/dL. APACHE II score was calculated to be 22, indicating 28.6% mortality. SLEDAI (systemic lupus erythematosus severity index) was calculated to be 29. The patient was treated as presumed catastrophic antiphospholipid syndrome. Initial treatment included six sessions of plasmapheresis without intravenous immunoglobulin to avoid further kidney damage, and one dose of cyclophosphamide. After six sessions of plasmapheresis, the patient showed significant clinical improvement with resolution of her abdominal pain. Kidney function also improved with creatinine returning to baseline. Her blood pressure was difficult control, but eventually stabilized on a combination of a beta blocker, a calcium channel blocker, and a loop diuretic. The patient was discharged 4 weeks after initial presentation on prednisone 30 mg once per day, metoclopramide 5 mg every 6 hours, clonidine 1 patch per week, nifedipine 60 mg twice per day, and hydroxychloroquine 400 mg once per day. A repeat abdominal CT scan 5 week after the acute event showed complete resolution of pancreatitis (Figure 3). The patient remains free of recurrence of ischemic pancreatitis at 16 month follow-up.
Figure 2. CT scan showing inflammatory changes around pancreas
with small amounts of peripancreatic fluid, thickening of stomach
all, and free fluid in the pelvis, all consistent with pancreatitis.
Figure 3. CT scan showing normal pancreas with no fluid
accumulation in the pelvis, and normal stomach wall.
DISCUSSION
Antiphospholipid antibodies have a high prevalence in the general population. Some estimates place their prevalence among asymptomatic, healthy subjects at 1 to 5%, and much more common in patients with systemic lupus erythematosus, with 30-40% prevalence [5]. Lupus anticoagulant antibodies are actually a heterogeneous set of antibodies that are identified by prolongation of coagulation cascade assays. Although they delay coagulation in the assay, lupus anticoagulant antibodies are often prothrombotic in vivo as well. Similarly, the anti-cardiolipin and anti-beta2- microglobulin antibodies are defined by their ability to bind phospholipids or phospholipid-binding proteins on immunoassays [2, 6].
However, in certain individuals, these antibodies are active and cause clinical manifestations, primarily thromboses and fetal loss. The clinical manifestations against the background of these antibodies give rise to the antiphospholipid syndrome [7]. This condition is typically manageable using standard anticoagulation therapy. However, in a subset of individuals with antiphospholipid syndrome, the thromboses are widespread and affect several organ systems simultaneously, leading to a high rate of mortality. This entity has been termed catastrophic antiphospholipid syndrome [8]. In catastrophic antiphospholipid syndrome, the thromboses tend to be in the form of thrombotic microangiopathy, in contradistinction to the predominantly venous thromboses seen in antiphospholipid syndrome. Definitive catastrophic antiphospholipid syndrome occurs when three or more organ systems are affected by thromboses in less than a week in patients who have tested positive for antiphospholipid antibodies twice at least six weeks apart. There should also be confirmation by histopathology of small vessel occlusion in at least one organ or tissue. Patients who meet most but not all criteria are labeled as having probable catastrophic antiphospholipid syndrome [9].
Our patient displayed many features of catastrophic antiphospholipid syndrome, given that she also has systemic lupus erythematosus. In addition to her pancreatitis and worsening kidney function, she had worsening hypertension that required multiple antihypertensive medications to manage. Given her clinical picture and our high clinical suspicion, we decided to treat her as if she had catastrophic antiphospholipid syndrome. Due to the aggressive nature of the syndrome, the high mortality rate, and the lag time in obtaining full laboratory results, treatment should be instituted as soon as the syndrome is suspected.
There are several treatment options for catastrophic antiphospholipid syndrome, but no definitive regimen. As the number of patients with catastrophic antiphospholipid syndrome worldwide is small, there are no prospective, randomized, controlled trials on therapeutic strategies [10]. First line therapies include anticoagulants and corticosteroids. Plasmapheresis and intravenous immunoglobulin are second line treatments. Given our patient’s history and lack of clinical response to anticoagulation, plasmapheresis was attempted. Other treatments have been described in the literature, such as the use of rituximab, an anti- CD20 monoclonal antibody that acts against B-cells [11, 12]. However, the use of these alternate modalities has been limited to a few small case series.
syndrome and systemic lupus erythematosus, it is interesting to consider how catastrophic antiphospholipid syndrome manifests itself in systemic lupus erythematosus patients. As Bayraktar et al. have shown, patients with catastrophic antiphospholipid syndrome in systemic lupus erythematosus are more likely to be female and younger, have cerebral and pancreatic involvement, receive corticosteroids and cyclophosphamide, demonstrate a lower prevalence of high IgG anti-cardiolipin, and have a higher risk for mortality after adjusting for age, sex, organ involvement, and treatment [13]. Patients with catastrophic antiphospholipid syndrome in systemic lupus erythematosus who were treated with a combination of plasmapheresis, corticosteroids, and anticoagulation had a survival rate of 65% [14]. Our patient fit these criteria fairly well, as she was female, young, had pancreatic involvement, and responded well to plasmapheresis.
CONCLUSION
This case reaffirms the heightened degree of clinical awareness required to treat patients who present with signs of organ failure and have a history of systemic lupus erythematosus and antiphospholipid syndrome. Fortunately for our patient, her clinical picture improved after six cycles of plasmapheresis, corticosteroids, and cyclophosphamide. The patient has not had recurrent pancreatitis for 18 months of followup. However, given her multiple comorbidities, it is important to retain a high degree of suspicion for ischemic complications and catastrophic antiphospholipid syndrome due to its high mortality rate.
Conflict of interest The authors have no potential conflicts of interest
References
- Levine JS, Branch DW, Rauch J. The Antiphospholipid Syndrome. N Engl J Med 2002;346:752-763. [PMID 11882732]
- Asherson RA, Cervera R, de Groot PG, Erkan D, Boffa M-C, Piette J-C, et al. Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classication criteria and treatment guidelines. Lupus 2003; 12:530-534. [PMID 12892393]
- Wang CR, Hseih HC, Lee GL, Chuang CY, Chen CY. Pancreatitis associated to antiphospholipid antibody syndrome in patient with systemic lupus erythematosis. J Rheumatol 1992; 19:1123-1125. [PMID 1512770]
- Yeh TS, Wang CR, Lee YT, Chuang CY, Chen CY. Acute pancreatitis related to anticardiolipin antibodies in lupus patients visiting an emergency department. Am J Emerg Med 1993; 11:230- 232. [PMID 8489664]
- Love PE, Santor SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and non-SLE disorders. Ann Intern Med 1990; 112: 682-698. [PMID 2110431]
- Asherson RA, Khamastha MA, Ordi- Ros J. Primary antiphospholipid syndrome: major clinical and serological features. Medicine 1989; 68: 366-374.
- Uthman I, Godeau B, Taher A et al. The hematologic manifestations of the antiphospholipid syndrome. Blood Rev 2008; 22: 187-194.
- Bortolati M, Marson P, Fabris F et al. Recovery from catastrophic antiphospholipid syndrome by a plasma exchange procedure: report of four cases and review of the literature. Autoimmun Rev 2009; 8: 297-301.
- Espinosa G, Bucciarelli S, Asherson RA et al. Morbidity and mortality in the catastrophic antiphospholipid syndrome: pathophysiology, causes of death, and prognostic factors. SeminThrombHemost 2008; 34: 290-4.
- Spencer HL. Primary antiphospholipid syndrome as a new cause of autoimmune pancreatitis. Gut 2004 Mar;53(3):468; author reply 468.
- Erkan D. Therapeutic and prognostic considerations in catastrophic antiphospholipid syndrome. Autoimmunity Reviews 2006; 6:98-103.
- Harris EN, Pierangeli SS. Primary, secondary, and catastrophic antiphospholipid syndrome: what’s in a name? SeminThrombHemost 2008; 34: 219-26.
- Rubenstein E, Arkfeld DG, Metyas S, Shinada S, Ehresmann S, Liebman HA. Rituximab Treatment for Resistant Antiphospholipid Syndrome. J Rheumatol 2006; 33(2):355-357.
- Bayraktar UD, Erkan D, Bucciarelli S, Espinosa G, Asherson R, Catastrophic Antiphospholipid Syndrome Project Group. The clinical spectrum of catastrophic antiphospholipid syndrome in the absence and presence of lupus. J Rheumatol 2007; 34(2):346-352.




