Keywords
c-FLIP; Apoptosis; Death receptor pathways; Cancer biomarker; Antiapoptotic proteins; Cytokine resistance; Drug resistance
Abbreviations:
FADD: Fas-Associated Via Death Domain; c-FLIP: Cellular FLICE (FADD-Like IL- 1β-Converting Enzyme)- Inhibitory Protein; AD: Alzheimer's Disease; COPD: Chronic Obstructive Pulmonary Disease; UPR: Unfolded Protein Response
Introduction
c-FLIP or CLARP (caspase-like apoptosis-regulatory protein) plays important roles in apoptosis, necroptotic cell death [1-4], and autophagy [1], and triggers resistance to anticancer agents and cytokines, and autophagy [1,5-10] as well as in immune microenvironment [1,9]. Furthermore, recent results show that c-FLIP plays important roles in other diseases [11-13]. Therefore, c-FLIP is a valuable biomarker of prognosis and a reliable molecular target for developing therapeutics for cancer [1,9,14,15], AD, and immune-related diseases. This review concentrates on the usefulness of c-FLIP as a biomarker. We discuss how c-FLIP prevents apoptosis and induces cytokine and chemotherapy drug resistance in cancer cells, discuss its role for cancer prognosis, necrosis and autophagy, and its involvement as a marker of AD and COPD.
Literature Review
Apoptosis signaling pathways and role of c-FLIP
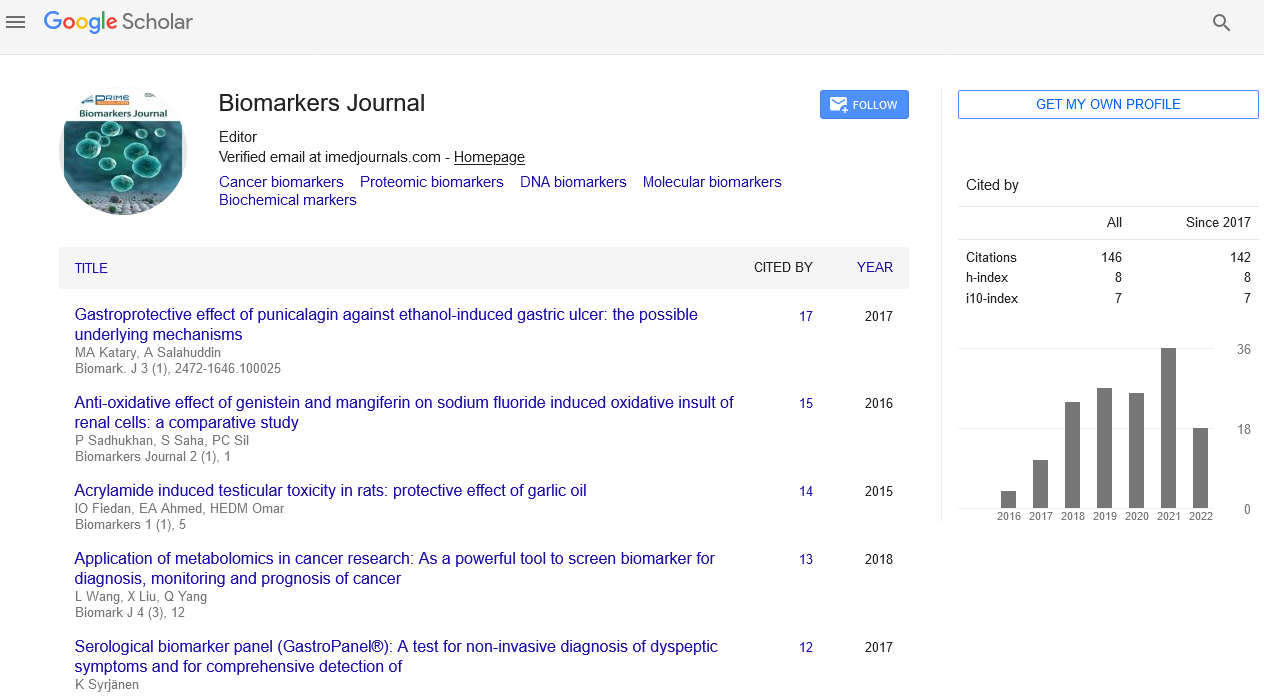
Three major signaling pathways, the intrinsic or mitochondrial pathway, the extrinsic or cell surface death receptors (DRs) pathways, and the endoplasmic reticulum (ER) stress-induced apoptosis pathway are known to regulate apoptosis (Figures 1 and 2) [1,4]. In the mitochondrial apoptotic pathway, anti- and proapoptotic members of the Bcl- 2 family cooperate and regulate the release of cytochrome c and other apoptosis-inducing factors from the mitochondria to the cytosol [16,17]. Cytochrome c and dATP bind to apoptotic proteinase-activating factor-1 (Apaf- 1) and make a complex with adenine nucleotides forms the apoptosome, which promotes procaspase-9 autoactivation [17,18]. Formation of apoptosome is important in the mitochondrial apoptosis pathway and by binding to cytochrome c activates the initiator caspase-9. This caspase in turn activates caspases-2, -3, -6, -7, -8, and -10 [17-22]. Successful apoptosis induced by requires direct activation of the pro-apoptotic proteins Bax and BAK at the mitochondria by a member of the pro-apoptotic family of proteins including Bid, Bim, or PUMA [20]. Cytochrome c, release and opening the permeability transition pore (PTP), and a collapse of mitochondrial transmembrane potential (Δψm) are associated processes and are due to the intake of endoplasmic reticulum (ER) Ca2+ release [20- 22] into cytosol.
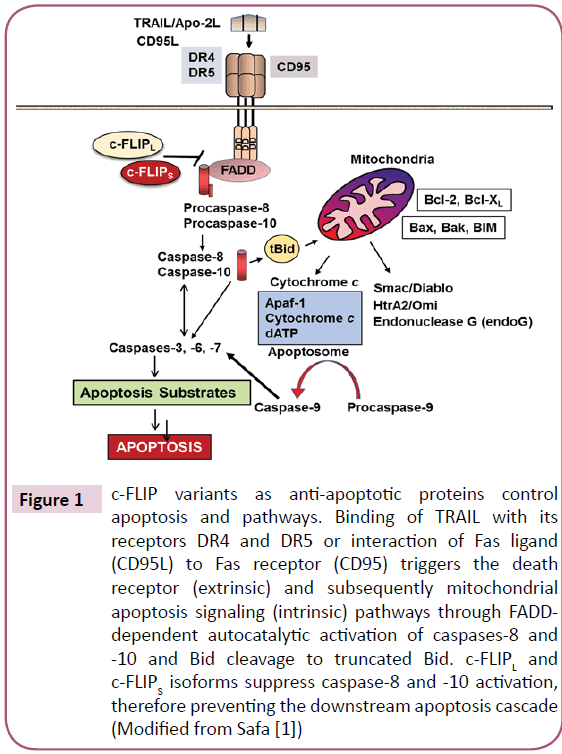
Figure 1: c-FLIP variants as anti-apoptotic proteins control apoptosis and pathways. Binding of TRAIL with its receptors DR4 and DR5 or interaction of Fas ligand (CD95L) to Fas receptor (CD95) triggers the death receptor (extrinsic) and subsequently mitochondrial apoptosis signaling (intrinsic) pathways through FADDdependent autocatalytic activation of caspases-8 and -10 and Bid cleavage to truncated Bid. c-FLIPL and c-FLIPS isoforms suppress caspase-8 and -10 activation, therefore preventing the downstream apoptosis cascade (Modified from Safa [1])
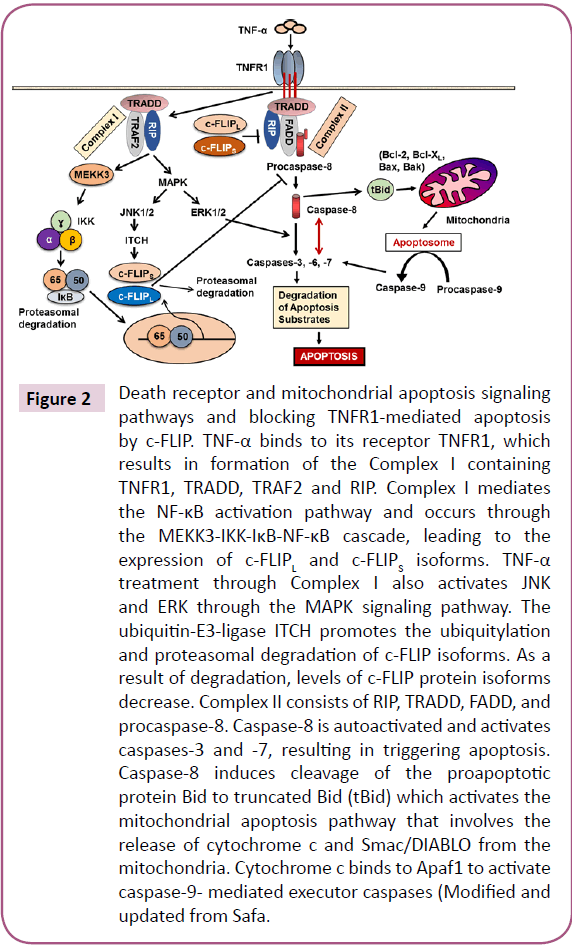
Figure 2: Death receptor and mitochondrial apoptosis signaling pathways and blocking TNFR1-mediated apoptosis by c-FLIP. TNF-α binds to its receptor TNFR1, which results in formation of the Complex I containing TNFR1, TRADD, TRAF2 and RIP. Complex I mediates the NF-κB activation pathway and occurs through the MEKK3-IKK-IκB-NF-κB cascade, leading to the expression of c-FLIPL and c-FLIPS isoforms. TNF-α treatment through Complex I also activates JNK and ERK through the MAPK signaling pathway. The ubiquitin-E3-ligase ITCH promotes the ubiquitylation and proteasomal degradation of c-FLIP isoforms. As a result of degradation, levels of c-FLIP protein isoforms decrease. Complex II consists of RIP, TRADD, FADD, and procaspase-8. Caspase-8 is autoactivated and activates caspases-3 and -7, resulting in triggering apoptosis. Caspase-8 induces cleavage of the proapoptotic protein Bid to truncated Bid (tBid) which activates the mitochondrial apoptosis pathway that involves the release of cytochrome c and Smac/DIABLO from the mitochondria. Cytochrome c binds to Apaf1 to activate caspase-9- mediated executor caspases (Modified and updated from Safa.
In the death receptor (DR)-mediated or extrinsic apoptosis pathway, Fas/Fas Ligand (FasL) [CD95/CD95 Ligand, CD95L] interaction, TRAIL/DR4 or TRAIL/DR5 interaction, or the binding of tumor necrosis factor α (TNF-α) with its receptor, TNF receptor 1 (TNFR1), initiates apoptosis (Figures 1 and 2). Similarly, binding of the agonistic antibodies to these respective receptors also initiates the apoptotic signaling cascade. Following the interactions of the DR ligands or DR agonistic antibodies with their respective trimerized DRs, the adaptor molecule Fas-Associated Death Domain (FADD) is recruited to the DRs via death domain (DD) interactions, whereas procaspase-8, procaspase-10, and c-FLIP are recruited to the deathinducing signaling complex (DISC) via death effector domain (DED) interactions [23,24]. Therefore, the DISC consists of trimerized DRs, FADD, procaspase-8/-10, and c-FLIP. Procaspase-8 and procaspase- 10 form a complex with FADD and autocatalytically activated to form the active initiator caspase-8 or caspase-10.
Caspase-8 also becomes activated intrinsically, and not extrinsically as a result of c-Myc inducing the down-regulation of c-FLIPL. Therefore, c-FLIPL may be of importance not only in regulating the death receptor ligand-induced apoptosis, but also in apoptotic processes triggered from within the cell [25]. Moreover, chromatin immunoprecipitation (ChIP) and luciferase assays identified the binding of c-Myc to the c-FLIPL promoter [26]. Moreover, after treating I/R rats with the c-Myc inhibitor 10058-F4, significant decrease in c-FLIPL and the increase in cleaved caspases-8 and 3 was seen, providing further support for the functional role of c-FLIPL in intrinsic apoptosis [26].
Silencing c-FLIP caused sensitivity of tumor cells to death ligands and chemotherapeutic agents in cancer cell models [6,27]. Furthermore, in addition to its function as an antiapoptotic protein, c-FLIP has other functions such as increased cell proliferation and tumorigenesis [3,27]. In TNF- α-triggered apoptosis (Figure 2), TNFR1 internalizes and induces formation of Complex II containing RIP, TRADD, FADD, and caspase-8. Caspase-8 autoactivation triggers activation of caspases-3 and -7, leading to apoptosis, and c-FLIP inhibits capsase-8 and -10 activation and apoptosis [3,28]. A pro-apoptotic Bcl-2 family member, Bid is cleaved to the truncated Bid (tBid) by caspase-8, and -10 and triggers mitochondrial cytochrome c release. Thus, tBid links the death receptor pathway to mitochondrial pathway. After activation, both caspases-8 and -9 activate caspase- 3 and other caspases, and ultimately apoptosis. Mitochondria play a leading role in cellular respiration and homeostasis in the cells and transfer various signals for cell survival and death to the cytosol.
Ranjan and Pathak [29] demonstrated that c-FLIPL and FADD expression participate in balancing redox potential by regulating antioxidant levels. Further, they noticed that knockdown of c-FLIPL and induced expression of FADD results in rapid accumulation of intracellular ROS accompanied by JNK1 activation to enhance apoptosis. Therefore, besides their death receptor signaling c-FLIPL and FADD play important roles in preventing mitochondrial mediated apoptosis. The interaction of the TNF-α trimer to TNF receptor 1 (TNFR1) also triggers TNFR1 trimerization and Complex I formation participating in inducing the antiapoptotic proteins (Figure 2). Complex I contains TNFR1, TRADD, TRAF2, and R IP and is able to activate the NF- κB signaling pathway through the MEKK3-IKK-IkB-NF-κB cascade and subsequently activates the transcription and expression of several genes including antiapoptotic factors such as IAPs, Bcl-2, and c-FLIP [3,30]. As shown in Figure 2, TNF-α treatment through Complex I can also cause activation of JNK and ERK through the MAPK signaling pathway. The ubiquitinspecific proteases system 2 (USP2) stabilizes the ubiquitin-E3-ligase ITCH and lowers NF-κB basal activity, which leads to reduced c-FLIP mRNA production; proteasomal degradation of c-FLIP isoforms is also elevated by its negative regulator proteasome ITCH [31]. Therefore, levels of c-FLIP protein isoforms decrease and apoptosis increases. The TRAIL receptors, DR4 or DR5, can also promote alternative signaling pathways such as JNK, MAPK, or NF-κB by recruiting RIP1 and TRAF2 or TRAF5 to form a secondary signaling complex [32,33]. Activation of NF-κB in this pathway also results in increased expression of c-FLIP (Figure 2). Studies with TRADDdeficient mouse embryo fibroblasts (MEFs) have documented that RIP1 is also recruited to the TRAIL receptor by interacting with TRADD, and both RIP1 and TRADD protect against TRAILinduced apoptosis [5,34]. In these TRADD-deficient MEFs, MAPK and NF-κB pathway activation was impaired, confirming the role of TRADD as the key adaptor protein mediating nonapoptotic signaling by DRs [5,34]. Indeed, the human T-cell leukemia virus type 1 (HTLV-1) Tax protein triggers c-FLIP production through activation of the IKK-NF-κB via DR4/DR5 pathway [35].
It is also known that TNF-α and Fas trigger the cleavage of mitogen-activated protein kinase/ERK kinase kinase (MEKK), resulting in production of a constitutive active form of MEKK1 and leading to JNK activation in c-FLIP knockdown cells [36]. In the absence of caspase-8 activity, the death receptors promotes death by programmed necrosis (necroptosis) which requires the kinases receptor-interacting kinase 1 (RIPK1), RIPK3, and mixedlineage kinase like protein (MLKL) [37].
The endoplasmic reticulum (ER) stress-induced apoptosis pathway is the result of internal cellular stress like the accumulation of damaged glycoproteins [38], or exogenous stress, such as chemotherapeutic agents and hypoxia [39], which activate the process of cellular responses to stress, termed the unfolded protein response (UPR) [40]. UPR plays a major role in the homeostasis of ER. Prolonged or excessive ER stress triggers signaling pathways resulting in cell death. Triggering apoptosis via ER stress is usually associated with increased expression of DR5, down regulation of c-FLIP, caspase activation, and participation o of the JNK pathway. ER and mitochondria cross-talk through forming mitochondria-ER associated membranes (MAMs) or mitochondria associated ER membranes (MERCs) [41] that reciprocally transmit signals under stress conditions, triggering synergistic death responses [42].
c-FLIP spliced variants and structure of c-FLIP isoforms
The mammalian cellular homolog of viral FLICE-inhibitory proteins (v-FLIPS) was termed c-FLIP [43]. While c-FLIP consists of a family of alternatively spliced variants, three of these variants including the 26 kDa short form (c-FLIPS), the 55 kDa long form (c-FLIPL) and the 24 kDa form (c- FLIPR) are expressed as proteins (Figure 3) [20,44]. c-FLIPR is smaller than c-FLIPS and has a similar pattern of expression as c-FLIPS during activation of primary human T cells and is strongly induced in T cells upon CD3/CD28 co-stimulation [45]. In humans, three isoforms of c-FLIP, c- FLIPL(55 kDa), c-FLIPS (27 kDa), and c-FLIPR (25 kDa) have been identified [1,46]. These isoforms contain two DEDs (Figure 3). c-FLIPLalso contains a catalytically inactive caspase- like [44] domains (p20 and p12). Moreover, c-FLIPS has an additional isoform specific 19 amino acids in its C-terminal domain. Moreover, c-FLIPL has a caspase-8 cleavage site at position Asp- 376 (LEVD) and cleavage at this site results in generation of the fragment variant p43c-FLIP, p43- FLIP, and p22-FLIP [1,47]. The C-terminal region of c-FLIPS and c-FLIPR plays an important role in ubiquitination and degradation and their anti-apoptotic function.
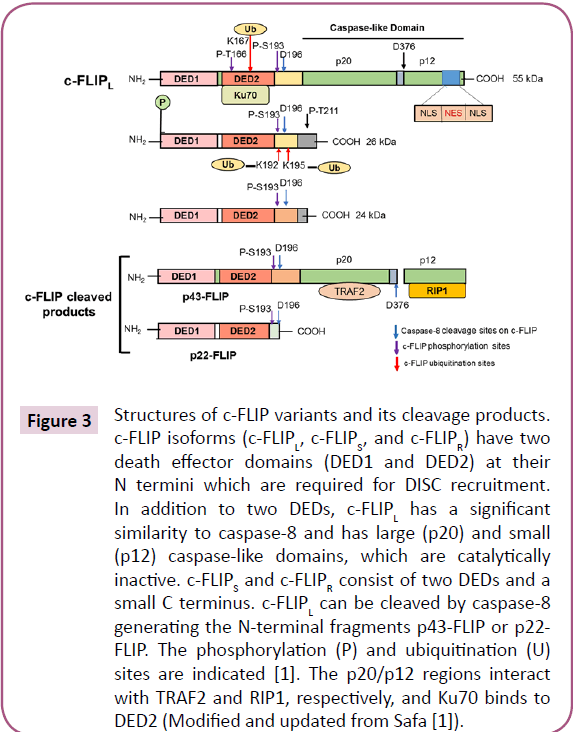
Figure 3: Structures of c-FLIP variants and its cleavage products. c-FLIP isoforms (c-FLIPL, c-FLIPS, and c-FLIPR) have two death effector domains (DED1 and DED2) at their N termini which are required for DISC recruitment. In addition to two DEDs, c-FLIPL has a significant similarity to caspase-8 and has large (p20) and small (p12) caspase-like domains, which are catalytically inactive. c-FLIPS and c-FLIPR consist of two DEDs and a small C terminus. c-FLIPL can be cleaved by caspase-8 generating the N-terminal fragments p43-FLIP or p22- FLIP. The phosphorylation (P) and ubiquitination (U) sites are indicated [1]. The p20/p12 regions interact with TRAF2 and RIP1, respectively, and Ku70 binds to DED2 (Modified and updated from Safa [1]).
c-FLIP activates cytoprotective and proliferation pathways
As we previously discussed [1] several pathways with major roles in regulating cell survival, proliferation, and carcinogenesis can be activated by c-FLIP (Figure 2) [47-55]. DNA-PK/Akt pathway is also can regulate the expression of c-FLIP [56]. Furthermore, interaction of c-FLIPL with MKK7 might selectively suppress JNK activation [57]. c-FLIP may also regulate pathways participating in the production of inflammatory cytokines, tumor cell migration and metastasis [58,59], alter cell cycle progression and enhance cell proliferation and carcinogenesis [1,9]. Furthermore, overexpression of c-FLIP can alter cell cycle progression and enhance cell proliferation and carcinogenesis [1,9].
c-FLIP as a an anti-apoptotic protein
It has been shown that c-FLIP inhibit death receptor-mediated apoptosis as well as apoptosis induced by a variety of cancer chemotherapeutic agents and small molecule targeted anticancer compounds and ionizing radiation [1,51]. Additionally, major roles for c-FLIP in promoting the survival of immunosuppressive tumor-promoting immune cells have been discovered [9]. Thus, c- FLIP is a rational anticancer therapeutic target. c-FLIPL modulates signals [11] leading to both cell death and cell growth [60]. c-FLIPL is upregulated in human tumors [51,61], rendering tumor cells resistant to therapies and immune surveillance [62], Regulatory T cells or Tregs turn off immune responses and have been used for immunotherapies. Treg cells compared to conventional T cells are more susceptible to apoptosis due to low c-FLIP expression [63]. Treg- specific deletion of c-FLIP in mice has been shown to cause fatal autoimmune disease [29]. Therefore, a c-FLIP function is essential for cell homeostasis and prevention of autoimmunity in Treg [63]. Given the fact that Treg cells control autoimmunity and inhibit anti-cancer immunity, c-FLIP could be used as a useful therapeutic target to modulate Treg cell abundance and immune responses in cancer or autoimmune disease [63].
c-FLIP has been shown to affect immune regulatory pathways enriched in NF-κB response to TNF, cytokine network, and genes upregulated by IL-6 via STAT3 [64]. This specific gene upregulation may be partially linked to NF-κB activation induced by the nuclear translocation of c-FLIP [64]. These authors uncovered a critically important dual role of c-FLIP in myeloid cells. In mature monocytes, cancer-induced c-FLIP expression promoted immune suppressive functions and increased survival. On the other hand, constitutive c-FLIP activation in the myeloid lineage induced chronic inflammation associated with myeloproliferation and immune suppression. These results revealed that c-FLIP emerged as a novel factor for controlling cancer-associated chronic inflammation and immune dysfunction.
Role of c-FLIP in programmed necroptosis
Necroptosis is physiologically regulated by specific proteins including RIPK1, receptor interacting protein kinase 3 (RIPK3, also known as RIP3), and mixed lineage kinase-like protein (MLKL) [1]. Receptor interacting protein kinase 1 (RIPK1), also known as RIP1, is a protein which interacts with the TNF-α receptor 1 signaling complex and recent advances have shown the critical roles of RIPK1 in cell survival, apoptosis, and necroptosis [65,66]. Necroptosis is induced as a result of inhibition of caspase-8 activation regulated by the death platform complex, Ripoptosome, and c-FLIP isoforms can switch apoptotic and necroptotic cell death. Moreover, the cellular inhibitor of apoptosis proteins (cIAPs) blocks Repotosome formation [1]. Interestingly, while c-FLIPL is able to prevent Ripoptosome formation, c-FLIPS promotes its formation [1]. Therefore, these c-FLIP isoforms in the Ripoptosome determine whether cell death occurs by RIP3- dependent necroptosis or caspase-dependent apoptosis [1,67,68]. Molecularly, RIPK1 phosphorylates and activates RIPK3, and activated RIPK3 then phosphorylates MLKL. Subsequently, the phosphorylated MLKL oligomer form translocates to the plasma membrane and induces necrotic cell death by forming pores on the plasma membrane [69].
Autophagy and c-FLIP
In addition to inhibiting apoptosis, c-FLIPL can affect autophagy by (1) direct action on the autophagy by competing with Atg3 binding to LC3, reducing LC3 processing and inhibiting autophagosome formation [1,70,71] and (2) by interacting with procaspase-10, it forms an enzymatic complex that can cleave the Bcl-2 associated transcription factor 1 (BCLAF1), which is an autophagy inducer. This cleaved BCLAF1 form displaces Bcl-2 from an inhibitory complex with Beclin-1 and Beclin-1-induced autophagy [71]. Disrupting or preventing formation of the procaspase-10/c-FLIPL complex may induce autophagic cell death [71]. Recently, it was shown that inhibition of c-FLIP overcomes acquired resistance to sorafenib by reducing endoplasmic reticulum stress (ERS)-related autophagy in hepatocellular carcinoma (HCC) [72].
c-FLIP as a valuable prognostic biomarker in various cancers
c-FLIP variants (c-FLIPL and c-FLIPS) serve as prognostic biomarkers for various cancer types. As shown in Figure 3, c-FLIPL has a bipartite nuclear localization signal (NLS) and a nuclear export signal (NES) in its C-terminal region, required for its transport between the nucleus [10] and the cytosol [73,74]. High expressions of c-FLIP are in lethal human cancers including ovarian, colon, cervical, glioblastoma, breast, colorectal, and prostate cancers, and multiple myeloma [1,50,51,61,75,76]. Collectively, c-FLIP expression is frequently upregulated in various malignancies and correlates with poor prognosis. Immunohistochemical analysis has shown two distinct pools of c-FLIPL, which correlate the expression and the subcellular localization of c-FLIPL protein with patient therapeutic and survival outcomes [75,76]. Humphreys et al. [9] in a cohort of 184 non-small lung cancer (NSCLC) patients demonstrated high cytoplasmic but not nuclear c-FLIP significantly correlated with decreased overall survival. Valnet- Rabier et al. [77] showed that in 32 Burkitt's lymphoma cases, a mainly cytoplasmic pool of c-FLIP was highly correlated with poor patient outcome [77]. Several studies have identified c-FLIP as an independent adverse indicator in cancer and found both c-FLIPL and c-FLIPS play important role in cancer patient treatment outcomes and that the expression of particular c-FLIP isoforms has prognostic clinical value [66,78-86]. Ullenhag et al. [87] showed in colorectal (CLR) cancer patients that c-FLIPL is an independent marker of poor prognosis. Furthermore, high c-FLIP level was shown to be an independent adverse prognostic marker in stage II and III CLR cancer and might identify patients with high possibility of relapse [78]. Moreover, c-FLIPL mRNA significantly correlates with poorer overall survival of a cohort of acute myeloid leukemia (AML) patients [88]. Zang et al. [89] using immunohistochemistry fund c-FLIP protein expression in IA2-IIIA cervical- squamous cell carcinoma patients and showed that high c-FLIP level was an independent negative indicator for diseasefree survival (p=0.015). Lee et al. [90] fund that c-FLIPL expression in invasive breast carcinomas with c-FLIPL-positive patients showing a poor prognosis (p<0.01). Furthermore, expression of c-FLIPL, lymph nodes status, and molecular subtypes were independent prognostic factors for these patients (p<0.05).
McCourt et al. [91] have reported that expression levels of c-FLIP and heat shock protein 27 (HSP27) in prostate cancer correlated with the Gleason score sum and pathologic stage. A Prostate cancer Gleason score or grade assists in determining how aggressively the tumor is likely to behave. The score helps to classify the cancer by grading how quickly it is likely to grow and is an indicator of how likely it is to metastasize outside the prostate gland. Elevated expression of c-FLIP was shown to antagonize the therapeutic response to androgen receptor targeted therapy in castration-resistant prostate cancer (CRPC) [92]. Furthermore, the overexpression of stromal c- FLIP promotes androgen-dependent prostate cancer growth and invasion [93]. Another significant role of c-FLIP is in the carcinogenesis and aggressiveness of endometrial carcinoma and might be a critical prognostic factor in this caner [94]. Acute human papillomavirus (HPV) infection causes cervical intraepithelial neoplasia marked by high copy episomal viral DNA and L1/L2 capsid protein expression in the cells that facilitate sexual viral transmission. Recently, Nuovo et al. [14] showed that increased level of c-FLIP and elevated expression of importin-β, exportin-5, Mcl1, p16, and Ki67 are new biomarkers of human papillomavirus infection in acute cervical intraepithelial neoplasia.
Identification of reliable biomarkers remains a crucial factor to evaluate clinical progress in pancreatic ductal adenocarcinoma (PDAC), a lethal cancer. Haag et al. [94] found that c-FLIP overexpression in pancreatic intraepithelial neoplasia (PanIN) lesions (the most prevalent type of early lesion, arising from the ductal epithelial cells leading to moderate dysplasia, highgrade dysplasia, and invasive carcinoma), and PDAC, compared to normal pancreatic ducts. In addition, knockdown of c-FLIP increased death receptor-triggered apoptosis in PDAC cell lines. Schmid et al. [95] demonstrated that c-FLIP expression status is a [32] valuable prognostic biomarker in PDAC. Subsequently, these investigators explored the prognostic significance of c- FLIP protein expression in PDAC in a well-defined cohort including clinical parameters, and other PDAC cohorts. Interestingly, the complete lack of c-FLIP was associated with a highly aggressive clinical course [94]. c-FLIPL is usually considered to function as an antiapoptotic protein [96]. However, it is also believed that c-FLIPL may also function as a pro-apoptotic protein [48], depending on its isoform and expression levels [48,96]. Indeed, it is known that high [19] expression of c-FLIPL blocks caspase-8 activation [27,96] while physiological L levels of c-FLIPL enhance oligomerization and autoproteolytic processing of caspase [27,96]. c-FLIPS has been implicated to function in antiapoptotic signaling as well as in increased cell death [48]. Interestingly, c-FLIPS has been shown to promote necroptosis in response to Toll-like receptor 3 stimulation and depletion of IAP proteins [68] by increasing the formation of a cytosolic complex containing RIP1, FADD, and caspase-8 [1,68]. Therefore, the finding that the absence of c-FLIP is an indicator of poor prognosis in PDAC may be due to concept that c-FLIPL can function both as an antiapoptotic and proapoptotic factor, dependent on the tumor environment. The absence of c-FLIP in PDAC as an independent indicator of short overall survival implies that c-FLIP has prognostic value. Moreover, further research into the prognostic relevance of c-FLIP in PDAC is required to validate the impact of this protein as a prognostic biomarker for this devastating disease. Increased level of c-FLIP expression is a frequent event in stomach carcinoma [81,97], and as McCourt et al. [91] suggested, stomach carcinoma cells in vivo may require c-FLIP expression to evade apoptosis and its expression is associated with tumor cell proliferation in this cancer [98]. Additionally, c-FLIP expression and its relationship with clinicopathologic features of melanoma have been actively explored [99]. c-FLIP was found to have an important role in aggressiveness of malignant melanoma and is a useful predicting prognostic marker for patients with this disease [99].
c-FLIP as a mediator of anticancer therapy resistance
Increased expression of c-FLIP in various tumor types is associated with the chemotherapeutic resistance and silencing of c-FLIP restores the proapoptotic signaling cascades efficiently to enhance chemo-sensitivity [1,12,46,61]. The association between expression of c-FLIP variants and poor prognosis relates to the fact that c-FLIP confers resistance to a number of anticancer agents [6,48,99-102] including sorafenib [103], the inhibitor of RAF-1 and class II receptor tyrosine kinase. c-FLIP triggers TRAIL resistance due to its increased expression level in various human tumors as well as in cancer stem cells (CSCs) from these tumors [104]. Furthermore, silencing c-FLIP expression or using c-FLIP inhibitors sensitizes tumor cells and CSCs to TRAIL and drugs like Taxol, doxorubicin, cisplatin, gemcitabine, etc. [46,100,104-106]. CSCs are also resistant to chemotherapy and radiotherapy and play a significant role in cancer recurrence [100]. Expression levels of c-FLIP isoforms were significantly higher in glioblastoma cancer stem cells (GSCs) than the entire GBM tumor cell population, and c-FLIP silencing in GSCs enhanced TRAIL and temozolomide (TMZ)- induced apoptosis [107]. It is known that breast cancer stem cells (BCSCs) mediate tumor recurrence and drive tumor metastasis [105,108]. Piggott et al. [108] reported a therapeutic approach to selectively eliminate BCSCs. They found that c-FLIP is upregulated in BCSCs from various breast cancer subtypes and that silencing of c-FLIP by its siRNA partially sensitizes these cells to the anticancer agent TRAIL. Significantly, their data demonstrated that BCSCs are sensitive to derepression of the proapoptotic pathway by TRAIL due to c-FLIP silencing, which results in an 80% reduction i n primary tumors, a 98% reduction in tumor metastases, and the loss of BCSC self-renewal.
Overall, c-FLIP can serve as a biomarker for detecting CSCs that are refractory to cell death, and inhibition of c-FLIP by pharmacological agents or genetic approaches may be a rational therapeutic strategy to increase the efficacy of anti-cancer agents and eliminate the apoptosis- resistant CSCs. Phenotypic plasticity is a new paradigm for understanding the origin of CSCs and the genesis of inter conversion between differentiated cells and CSCs, which acquire self-renewal, proliferation, and resistance to therapy [109,110]. Recently, Thakur and Ray [111] identified NF- κB as a regulator of dedifferentiation, which increases both TNF-α and PIK3CA expression only in the enriched side-population (SP) containing CSCs, but not in the nonside-population in platinumresistant ovarian cancer cells treated with cisplatin. Activation of PI3K/AKT signaling pathway drives SPs into an undifferentiated state, through increased c-FLIP, P21, and P27 expression [111]. Interestingly, Piggott et al. [112] recently identified a novel mechanism of acquired vulnerability to an extrinsic apoptosis stimulus in breast cancer primary cultures and BCSCs with acquired resistance to tamoxifen (TAMR) or Faslodex (fulvestrant), which has both therapeutic and prognostic potential for breast cancer therapy. In parallel with developing an endocrine resistance phenotype, these cells acquired TRAIL sensitivity, which correlated with decreased expression of intracellular levels of c-FLIP and an increase in JNK-mediated phosphorylation of the E3-ligase ITCH, which degrades c-FLIP. Furthermore, while the apoptosis inducing agent lapatinib has clinical efficacy in the treating trastuzumab-refractory HER2-positive breast cancers, significant proportions of patients develop acquired resistance to the drug and develop progressive disease. Eustace et al. [113] demonstrated that the development of acquired resistance to lapatinib resulted in triggering TRAIL sensitivity. Mechanistically, increased sensitivity to TRAIL in these cells was related to decreased phosphorylation of AKT, elevated lenel of FOXO3a and reduced expression of c-FLIP [113]. c-FLIPL and c-FLIPS display several roles in various cellular signaling pathways, and activate and/or upregulate several cytoprotective and prosurvival signaling proteins that include Akt, ERK, and Wnt (Figure 4). Strategies to develop new cancer therapeutics that improve the efficacy and decrease the toxicity of chemotherapeutic agents by targeting specific c-FLIP isoforms is very attractive [48]. The association between c-FLIP expression and poor prognosis in tumors may be because c-FLIP confers resistance to several anticancer agents [46,100,104,106]. We have reported that increased expression of c-FLIPL or c-FLIPS triggers resistance to Taxol by interacting with caspases-8 and -10 and silencing c-FLIP isoforms increase Taxol-induced apoptosis in malignant cells [6]. In colorectal cancer (CRC), high c-FLIP expression triggered resistance to CRC standard-of-care chemotherapeutic agents, 5-fluorourcil-and oxaliplatin-triggered apoptosis both in vitro and in vivo [114]. Moreover, siRNA mediated silencing of c-FLIP isoforms (particularly c-FLIPL) significantly increase cell death by these drugs. Similarly, cisplatin in NSCLC models, Taxol in leukemia [6], anticancer drugs in renal cancer cells [106] and ionizing radiation in NSCLC [114] trigger more apoptosis when c-FLIP was silenced or inhibited by pharmacological inhibitors [1,46,85,101,114]. Interestingly, high expression of cytoplasmic c-FLIP is indicative of poor prognosis [75]. Recent data also show that inhibition of the bromodomain and extra terminal domain (BET) family inhibitors effectively inhibit c-FLIP expression and sensitize KRAS-mutated NSCLC cells to pro-apoptotic agents TRAIL and cisplatin [115].
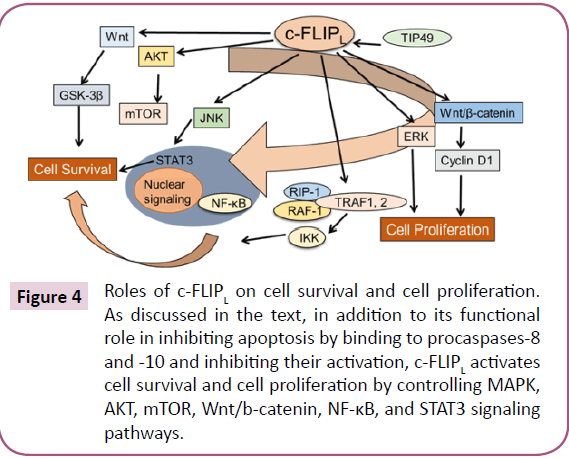
Figure 3: Roles of c-FLIPL on cell survival and cell proliferation. As discussed in the text, in addition to its functional role in inhibiting apoptosis by binding to procaspases-8 and -10 and inhibiting their activation, c-FLIPL activates cell survival and cell proliferation by controlling MAPK, AKT, mTOR, Wnt/b-catenin, NF-κB, and STAT3 signaling pathways.
Discussion
c-FLIP and the tumor immune microenvironment
c-FLIP effects on immune effector cells is a potential reason for the correlation between c-FLIP expression and poor prognosis in tumors with high c-FLIP expression. c-FLIP can prevent cell death induced by immune effector cells [63]. In fact, cytotoxic T lymphocytes (CTLs) and natural killer cells express Fas ligand (FasL) and TRAIL, but may exert immune resistance in [75] tumors with high c-FLIP expression. Overexpression of c-FLIP has been shown to allow establishment of tumors in immune-competent mice by blocking Fas-dependent cell death triggered by CTLs on their target tumor cells [62]. c-FLIP is also important in the survival of monocytic myeloid-derived suppressor cells (MDSCs) through its ability to inhibit death receptor mediated extrinsic apoptosis [12,62,116]. These MDSCs are immunosuppressive cells that are recruited to tumors [117] and that MDSC numbers may be a marker for poor prognosis in cancer patients [118,119]. Zhao et al. [120] have shown that signaling of TNFR-2, but not TNFR-1, promoted MDSC survival through up regulation of c-FLIP. These results demonstrate that TNFR-2 signaling promotes MDSC survival and accumulation through c-FLIP overexpression and helps tumor cells evade the [13] immune system [120]. Furthermore, c-FLIP is important for FoxP3+ regulatory T cells (Tregs) survival for maintaining local immunosuppressive environments [64]. Particularly significant is that c-FLIP knockout in dendritic cells enhanced production of TNF-α, IL2, or granulocyte-macrophage colony-stimulating factor (GM-CSF), in response to stimulation of TLR4, TLR2, and dectin-1 and increased T cell activation [121]. Therefore, c-FLIP [64] has a functional role in immunosuppression.
c-FLIP in Alzheimer’s disease (AD)
Abnormal c-FLIP protein expression has been identified in several diseases including multiple sclerosis (MS), Alzheimer's disease (AD), diabetes mellitus, rheumatoid arthritis (RA) and various cancers [30]. Fossati et al. [122] have fund that the TRAIL receptors DR4 and DR5 specifically mediate oligomeric amyloid-β (Aβ) induction of apoptotic in human microvascular cerebral endothelial cells through caspases-8 and -9 activation. Direct binding assays using receptor chimeras have confirmed the specific interaction of oligomeric Aβ with DR4 and DR5. DR4 and DR5 upregulation and colocalization with Aβ at the cell membrane suggests their involvement as initiators of the apoptotic cascade [122]. Aβ induced c-FLIP downregulation and the caspase- 8-triggered mitochondrial pathway of apoptosis through engagement of cleaved Bid. Conversely, apoptosis protection achieved through siRNA silencing of DR4 and DR5 highlighted their active role downstream apoptotic pathways unveiling new targets in (i.e., c-FLIP, DR4, and DR5) for therapeutic intervention for AD. Chemicals and miRNAs known to upregulate [77] c-FLIP may be used to relieve depression and anxiety among AD patients. The antidepressant Fluoxetine upregulates expression of c-FLIP through activating the c-FLIP promoter region spanning nucleotides -414 to -133, including the CREB and SP1 sites, and inhibits LPS-induced apoptosis in hippocampus-derived neural stem cells (NSCs) [123,124]. Moreover, fluoxetine treatment significantly inhibited the induction of proinflammatory factor IL-1β, IL-6, and TNF-α in the culture medium of LPS-treated NSCs. Therefore, the activation of c-FLIP by fluoxetine indicates its role in neuroprotection. Recent results show that fluoxetine could activate the Wnt/β-catenin signaling pathway and reduce amyloidosis in AD brain tissue [125]. Interestingly, the Wnt/β- catenin is activated by c-FLIP [124]. Therefore, c-FLIP can either directly or indirectly may be involved in initiating and maintaining AD. Accumulation of β-amyloid and hyperphosphorylated tau protein in AD is probably triggers the neurofibrillary tangles and plaques in this disease [123]. At the present, there is no histologic marker for AD and recent evidence suggests that the he hyperphosphorylation of tau protein in neurons may be [9,36] critical in AD. Previously, miR-512 has been shown to prevent c-FLIP expression and enhance Taxol triggered apoptosis in hepatocellular carcinoma cells [125]. Corroborating these results, miR-512 significantly is reduced in AD brain sections with high level of hyperphosphorylated tau protein. Interestingly, immunohistochemistry documented that the c-FLIP and MCL1, the 2 targets of miRNA-512, were significantly upregulated in AD brain, were colocalized with the abnormal tau protein, and no apoptosis was noted in these areas [12]. Therefore, these results suggest that increased expression of c-FLIP and MCL1 may change the balance between apoptosis and anti-apoptotis events in neurons. Similarly, using neuronal cell cultures, Nuovo et al. [11] suggested that molecular changes including accumulation of MCL1 and c-FLIP in the affected neurons in AD prevent cell to die and accumulate hyperphosphorylated tau and β-amyloid. There is no authentic histologic marker for AD's and the above data underscore the significance of MCL1 and c-FLIP as biomarkers for this disease and may offer clues to its etiology. Strategies to upregulate these proteins in Alzheimer's disease may help to identify agents to prevent or inhibit progress of this severe disease.
c-FLIP as a biomarker of COPD
Cigarette smoking (CS) is a critical risk factor for COPD [125]. Pouwels et al. [126] fund the c-FLIP gene, CFLAR is has a relationship with CS-triggered release of damage- associated molecular patterns (DAMP) in airway epithelial cells. Recently, Faiz et al. [13] studied the effect of CS on CFLAR expression levels in bronchial biopsies from smokers and non-smokers and isoform-expression of CFLAR transcript in air-liquid interface-differentiated bronchial epithelial cells (BECs). These investigators concluded that CS exposure significantly decreased CFLAR expression in BECs. Furthermore, there was a shift in relative levels of the isoforms c-FLIPS and c-FLIPL transcripts in bronchial biopsies of smokers compared to non-smokers, correlated with cell death. The proof of concept came from in vitro downregulation of CFLAR by siRNA, which showed increased apoptosis, necrosis, and DAMP release as observed in CS. As this study concluded, CS exposure downregulates CFLAR expression. Moreover, decrease in c-FLIP might increase susceptibility of BECs to immunogenic cell death. Therefore, c-FLIP serves as a molecular biomarker for DAMPs and COPD [127-129].
Conclusion
Disregulation of c-FLIP plays crucial roles in several diseases including cancer, AD, and COPD. c-FLIP is a master apoptosis regulator frequently overexpressed in various malignancies and its upregulation is often correlated with a poor prognosis. It is well documented that c-FLIP isoforms induce resistance to death receptor ligands such as TRAIL, chemotherapeutic agents, and various cell death mechanisms in malignant cells, and is a rational target for modulating therapy- resistant human malignances Transitory pharmacological inhibition of c-FLIP is adequate to sensitize cancer cells to chemotherapeutic. Agents that indirectly target c-FLIP, such as drug entinostat (also known as MS-275 or SNDX-275) used for aromatase inhibitor?resistant breast cancer, have therapeutic window and usefulness, which suggests more specific c-FLIP?targeted agents will also be tolerated and effective. Moreover, in addition to high expression of c-FLIP in various cancers, specific mutations may serve as biomarkers and provide c-FLIP “dependency” in certain cancers. For example, an important function of c-FLIPL is activation of the NLRP3 and AIM2 inflammasomes and NSCLC cells with NLRP3 mutations are hypersensitive to cell death due to the loss of c-FLIP expression. Therefore, this hypersensitivity serves as a potential biomarker to identify a subgroup of patients who display enhanced vulnerability to c-FLIP?targeted therapies. Significantly, c-FLIP plays major roles in promoting the survival of immunosuppressive tumor promoting immune cells and function in inflammation, Alzheimer's disease and COPD. Thus, the foregoing discussion and these conclusions show that c-FLIP can serve as a versatile biomarker for cancer prognosis, a diagnostic marker for several diseases, and as a therapeutic window for direct targeting.
Acknowledgment
We would like to thank Dr. Mary D. Kraeszig for her editorial assistance. The work in the author’s laboratory on drug resistance and apoptosis signaling pathways was supported by research grants from the National Cancer Institute (CA 080734, CA 90878, and CA 101743), and Department of Defense (DOD) (OC 06095).
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- Safa AR (2013) Roles of c-FLIP in apoptosis, necroptosis, and autophagy. J Carcinog Mutagen 6: 3.
- Cain K, Bratton SB, Langlais C, Walker G, Brown DG, et al. (2000) Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J Biol Chem 275: 6067-6070.
- Bagnoli M, Canevari S, Mezzanzanica D (2010) Cellular FLICE inhibitory protein (c-FLIP) signalling: A key regulator of receptor-mediated apoptosis in physiologic context and in cancer. Int J Biochem Cell Biol 42: 210-213.
- Yu JW, Jeffrey PD, Shi Y (2009) Mechanism of procaspase-8 activation by c-FLIPL. Proc Natl Acad Sci USA 106: 8169-8174.
- Cao X, Pobezinskaya YL, Morgan MJ, Liu ZG (2011) The role of TRADD in TRAIL-induced apoptosis and signaling. FASEB J 4: 1353-1358.
- Day TW, Najafi F, Wu CH, Safa AR (2006) Cellular FLICE-like inhibitory protein (c-FLIP): A novel target for Taxol induced apoptosis. Biochem Pharmacol 71: 1551-1561.
- Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, et al. (2018) Endoplasmic reticulum stress signaling-from basic mechanisms to clinical applications. FEBS J 286: 241-278.
- Pfeffer CM, Singh ATK (2018) Apoptosis: A target for anticancer therapy. Int J Mol Sci 19: E448.
- Humphreys L, Espona-Fiedler M, Longley DB (2018) FLIP as a therapeutic target in cancer. FEBS J 285: 4104-4123.
- Perez LE, Parquet N, Shain K, Nimmanapalli R, Alsina M, et al. (2008) Bone marrow stroma confers resistance to Apo2 ligand/TRAIL in multiple myeloma in part by regulating c-FLIP. J Immunol 180: 1545-1555.
- Nuovo G, Paniccia B, Mezache L, Quiñónez M, Williams J, et al. (2017) Diagnostic pathology of Alzheimer's disease from routine microscopy to immunohistochemistry and experimental correlations. Ann Diagn Pathol 28: 24-29.
- Mezache L, Mikhail M, Garofalo M, Nuovo GJ (2015) Reduced miR-512 and the elevated expression of its targets cFLIP and MCL1 localize to neurons with hyperphosphorylated Tau protein in Alzheimer disease. Appl Immunohistochem Mol Morphol 23: 615-623.
- Faiz A, Heijink IH, Vermeulen CJ, Guryev V, Berge MVD, et al. (2018) Cigarette smoke exposure decreases CFLAR expression in the bronchial epithelium, augmenting susceptibility for lung epithelial cell death and DAMP release. Sci Rep 8: 124-126.
- Nuovo GJ, De-Andrade CV, Wells SI, Brusadelli M, Nicol AF (2018) New biomarkers of human papillomavirus infection in acute cervical intraepithelial neoplasia. Ann Diagn Pathol 36: 21-27.
- Li X, Pan X, Zhang H, Lei D, Liu D, et al. (2008) Overexpression of c-FLIP in head and neck squamous cell carcinoma and its clinicopathologic correlations. J Cancer Res Clin Oncol 134: 609-615.
- Grimm S (2012) The ER-mitochondria interface: The social network of cell death. Biochem Biophys Acta 1823: 327-334.
- Shakeri R, Kheirollahi A, Davoodi J (2017) Apaf-1: Regulation and function in cell death. Biochimie 135: 111-125.
- Edlich F (2018) BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem Biophys Res Commun 500: 26-34.
- Aouacheria A, Baghdiguian S, Lamb HM, Huska JD, Pineda FJ, et al. (2017) Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem Int 109: 141-161.
- Vela L, Marzo I (2015) Bcl-2 family of proteins as drug targets for cancer chemotherapy: The long way of BH3 mimetics from bench to bedside. Curr Opin Pharmacol 23: 74-81.
- Correia C, Lee SH, Meng XW, Vincelette ND, Knorr KL, et al. (2015) Emerging understanding of Bcl-2 biology: implications for neoplastic progression and treatment. Biochem Biophys Acta 1853: 1658-1671.
- Krammer PH, Kami?ski M, Kiessling M, Gülow K (2007) No life without death. Adv Cancer Res 97: 111-138.
- Lavrik IN, Krammer PH (2012) Regulation of CD95/Fas signaling at the DISC. Cell Death Differ 19: 36-41.
- Lavrik I, Golks A, Krammer PH (2005) Death receptor signaling. J Cell Sci 118: 265-267.
- Järvinen K, Hotti A, Santos L, Nummela P, Hölttä E (2011) Caspase-8, c-FLIP, and caspase- 9 in c-Myc-induced apoptosis of fibroblasts. Exp Cell Res N 317: 2602-2615.
- Xu D, Wang B, Chen PP, Wang YZ, Miao NJ, et al. (2018) c-Myc promotes tubular cell apoptosis in ischemia-reperfusion-induced renal injury by negatively regulating c-FLIP and enhancing FasL/Fas-mediated apoptosis pathway. Acta Pharmacol Sin 2: 1.
- Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, et al. (2002) c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J 21: 3704-3714.
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 86: 147-157.
- Ranjan K, Pathak C (2016) Expression of FADD and cFLIPL balances mitochondrial integrity and redox signaling to substantiate apoptotic cell death. Mol Cell Biochem 422: 135-150.
- Micheau O (2003) Cellular FLICE-inhibitory protein: An attractive therapeutic target?. Expert Opin Ther Targets 7: 559-573.
- Haimerl F, Erhardt A, Sass G, Tiegs G (2009) Down-regulation of the de-ubiquitinating enzyme ubiquitin-specific protease 2 contributes to tumor necrosis factor-alpha-induced hepatocyte survival. J Biol Chem 284: 495-504.
- Yang A, Wilson NS, Ashkenazi A (2010) Proapoptotic DR4 and DR5 signaling in cancer cells: Toward clinical translation. Curr Opin Cell Biol 22: 837-844.
- Trivedi R, Mishra DP (2015) Trailing TRAIL resistance: Novel targets for TRAIL sensitization in cancer cells. Front Oncol 5: 69.
- Füllsack S, Rosenthal A, Wajant H, Siegmund D (2019) Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell Death Dis 10: 122.
- Wang W, Zhou J, Shi J, Zhang Y, Liu S, et al. (2014) Human T-cell leukemia virus type 1 Tax-deregulated autophagy pathway and c-FLIP expression contribute to resistance against death receptor-mediated apoptosis. J Virol 88: 2786-2798.
- Nakajima A, Kojima Y, Nakayama M, Yagita H, Okumura K, et al. (2008) Downregulation of c-FLIP promotes caspase-dependent JNK activation and reactive oxygen species accumulation in tumor cells. Oncogene 27: 76-84.
- Silke J, Strasser A (2013) The FLIP side of life. Sci Signal 6: 2.
- Sano R, Reed JC (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833: 3460-3470.
- Wang M, Law ME, Castellano RK, Law BK (2018) The unfolded protein response as a target for anticancer therapeutics. Crit Rev Oncol Hematol 127: 66-79.
- Rah B, Nayak D, Rasool R, Chakraborty S, Katoch A, et al. (2016) Reprogramming of molecular switching events in UPR driven ER stress: Scope for development of anticancer therapeutics. Curr Mol Med 16: 690-701.
- Giacomello M, Pellegrini L (2016) The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ 23: 1417-1427.
- Marchi S, Patergnani S, Pinton P (2014) The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim Biophys Acta 1837: 461-469.
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388: 190-195.
- Ueffing N, Keil E, Freund C, Kühne R, Schulze-Osthoff K, et al. (2008) Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ 15: 773-782.
- Telieps T, Ewald F, Gereke M, Annemann M, Rauter Y, et al. (2013) Cellular-FLIP, Raji isoform (c-FLIPR) modulates cell death induction upon T-cell activation and infection. Eur J Immunol 43: 1499-1510.
- Safa AR, Pollok KE (2011) Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers (Basel) 3: 1639-1671.
- Oztürk S, Schleich K, Lavrik IN (2012) Cellular FLICE-like inhibitory proteins (c-FLIPs): Fine- tuners of life and death decisions. Exp Cell Res 318: 1324-1331.
- Safa AR, Day TW, Wu CH (2008) Cellular FLICE-like inhibitory protein (c-FLIP): A novel target for cancer therapy. Curr Cancer Drug Targets 8: 37-46.
- Safa AR (2012) c-FLIP: A master anti-apoptotic regulator. Exp Oncol 34: 176-184.
- Yang JK (2008) FLIP as an anti-cancer therapeutic target. Yonsei Med J 49: 19-27.
- Fulda S (2013) Targeting c-FLICE-like inhibitory protein (CFLAR) in cancer. Expert Opin Ther Targets 17: 195-201.
- Seidelin JB, Coskun M, Vainer B, Riis L, Soendergaard C (2013) ERK controls epithelial cell death receptor signaling and cellular FLICE-like inhibitory protein (c-FLIP) in ulcerative colitis. J Mol Med (Berl) 91: 839-849.
- Kataoka T, Tschopp J (2004) N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol 24: 2627-2636.
- Yu JW, Shi Y (2008) FLIP and the death effector domain family. Oncogene 27: 6216-6227.
- Gilot D, Serandour AL, Ilyin GP, Lagadic-Gossmann D, Loyer P, et al. (2005) A role for caspase-8 and c-FLIPL in proliferation and cell-cycle progression of primary hepatocytes. Carcinogenesis 26: 2086-2094.
- Kim MJ, Kim HB, Bae JH, Lee JW, Park SJ (2009) Sensitization of human K562 leukemic cells to TRAIL-induced apoptosis by inhibiting the DNA-PKcs/Akt-mediated cell survival pathway. Biochem Pharmacol 78: 573-582.
- Nakajima A, Komazawa-Sakon S, Takekawa M, Sasazuki T, Yeh WC (2006) An antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits the JNK pathway. EMBO J 25: 5549-5559.
- Naito M, Katayama R, Ishioka T, Suga A, Takubo K, et al. (2004) Cellular FLIP inhibits beta- catenin ubiquitylation and enhances Wnt signaling. Mol Cell Biol 24: 8418-8427.
- Leverkus M, Diessenbacher P, Geserick P (2008) FLIP ing the coin?: Death receptor- mediated signals during skin tumorigenesis. Exp Dermatol 17: 614-622.
- Lens SM, Kataoka T, Fortner KA, Tinel A, Ferrero I, et al. (2002) The caspase 8 inhibitor c- FLIP(L) modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol Cell Biol 22: 5419-5433.
- Shirley S, Micheau O (2013) Targeting c-FLIP in cancer. Cancer Lett 332: 141-150.
- Medema JP, De-Jong J, Van-Hall T, Melief CJ, Offringa R (1999) Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J Exp Med 190: 1033-1038.
- Plaza-Sirvent C, Schuster M, Neumann Y, Heise U, Pils MC, et al. (2017) c-FLIP expression in Foxp3-expressing cells is essential for survival of regulatory T cells and prevention of autoimmunity. Cell Rep 18: 12-22.
- Fiore A, Ugel S, De-Sanctis F, Sandri S, Fracasso G, et al. (2018) Induction of immunosuppressive functions and NF-κB by FLIP in monocytes. Nat Commun 9: 5193.
- Dhuriya YK, Sharma D (2018) Necroptosis: A regulated inflammatory mode of cell death. J Neuroinflammation 15: 199.
- Wang T, Jin Y, Yang W, Zhang L, Jin X, et al. (2017) Necroptosis in cancer: An angel or a demon?. Tumor Biology 39: 1-11.
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, et al. (2011) The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43: 432-448.
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, et al. (2011) cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by c-FLIP isoforms. Mol Cell 43: 449-463.
- Zhang J, Yang Y, He W, Sun L (2016) Necrosome core machinery: MLKL. Cell Mol Life Sci 73: 2153-2163.
- Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, et al. (2009) FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 11: 1355-1362.
- Lamy L, Ngo VN, Emre NCT, Shaffer AL, Yang Y, et al. (2013) Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 23: 435-449.
- Liu D, Fan Y, Li J, Cheng B, Lin W, et al. (2018) Inhibition of c-FLIP overcomes acquired resistance to sorafenib via reducing ER stress-related autophagy in hepatocellular carcinoma. Oncol Rep 40: 2206-2214.
- Zhang J, Chen Y, Huang Q, Cheng W, Kang Y, et al. (2009) Nuclear localization of c-FLIP- L and its regulation of AP-1 activity. Int J Biochem Cell Biol 41: 1678-1684.
- Katayama R, Ishioka T, Takada S, Takada R, Fujita N, et al. (2010) Modulation of Wnt signaling by the nuclear localization of cellular FLIP?L. J Cell Sci 123: 23-28.
- Riley JS, Hutchinson R, McArt DG, Crawford N, Holohan C, et al. (2013) Prognostic and therapeutic relevance of FLIP and procaspase?8 overexpression in non?small cell lung cancer. Cell Death Dis 4: e951.
- Yao Q, Du J, Lin J, Luo Y, Wang Y, et al. (2016) Prognostic significance of TRAIL signalling molecules in cervical squamous cell carcinoma. J Clin Pathol 69: 122-127.
- Valnet?Rabier MB, Challier B, Thiebault S, Angonin R, Margueritte G, et al. (2005) c?FLIP protein expression in Burkitt's lymphomas is associated with a poor clinical outcome. Br J Haematol 128: 767-773.
- McLornan DP, Barrett HL, Cummins R, McDermott U, McDowell C, et al. (2010) Prognostic significance of TRAIL signaling molecules in stage II and III colorectal cancer. Clin Cancer Res 16: 3442-3451.
- Chen LC, Chung IC, Hsueh C, Tsang NM, Chi LM, et al. (2010) The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ 17: 1463-1473.
- Zhou XD, Yu JP, Liu J, Luo HS, Chen HX, et al. (2004) Overexpression of cellular FLICE? inhibitory protein (FLIP) in gastric adenocarcinoma. Clin Sci 106: 397-405.
- Lee SH, Kim HS, Kim SY, Lee YS, Park WS, et al. (2003) Increased expression of FLIP, an inhibitor of Fas?mediated apoptosis, in stomach cancer. APMIS 111: 309-314.
- Tirapelli DP, Menezes SB, Franco IM, Lustosa IL, Rodrigues AR, et al. (2017) High expression of anti?apoptotic genes in grade I and II meningiomas. Arq Neuropsiquiatr 75: 209-215.
- Korkolopoulou P, Goudopoulou A, Voutsinas G, Thomas?Tsagli E, Kapralos P, et al. (2004) c?FLIP expression in bladder urothelial carcinomas: Its role in resistance to Fas?mediated apoptosis and clinicopathologic correlations. Urology 63: 1198-1204.
- Bagnoli M, Ambrogi F, Pilotti S, Alberti P, Ditto A, et al. (2009) c?FLIPL expression defines two ovarian cancer patient subsets and is a prognostic factor of adverse outcome. Endocr Relat Cancer 16: 443-453.
- McLaughlin KA, Nemeth Z, Bradley CA, Humphreys L, Stasik A, et al. (2016) FLIP: A targetable mediator of resistance to radiation in non-small cell lung cancer. Mol Cancer Ther 15: 2432-2441.
- Abedini MR, Qiu Q, Yan X, Tsang BK (2004) Possible role of FLICE?like inhibitory protein (FLIP) in chemo-resistant ovarian cancer cells in vitro. Oncogene 23: 6997-7004.
- Ullenhag GJ, Mukherjee A, Watson NF, Al-Attar AH, Scholefield JH, et al. (2007) Overexpression of FLIPL is an independent marker of poor prognosis in colorectal cancer patients. Clin Cancer Res 13: 5070-5075.
- McLornan D, Hay J, McLaughlin K, Holohan C, Burnett AK, et al. (2013) Prognostic and therapeutic relevance of c-FLIP in acute myeloid leukaemia. Br J Haematol 160: 188-198.
- Zang F, Wei X, Sun B (2014) Relationship of c-FLIPL protein expression with molecular subtyping and clinical prognosis in invasive breast cancer. Zhonghua Bing Li Xue Za Zhi 43: 442-446.
- Lee SW, Cho JM, Cho HJ, Kang JY, Kim EK, et al. (2015) Expression levels of heat shock protein 27 and cellular FLICE-like inhibitory protein in prostate cancer correlate with Gleason score sum and pathologic stage. Korean J Urol 7: 505-514.
- McCourt C, Maxwell P, Mazzucchelli R, Montironi R, Scarpelli M (2012) Elevation of c- FLIP in castrate-resistant prostate cancer antagonizes therapeutic response to androgen receptor-targeted therapy. Clin Cancer Res 18: 3822-3833.
- Ye H, Li Y, Melamed J, Pearce P, Wei J, et al. (2009) Stromal anti-apoptotic androgen receptor target gene c-FLIP in prostate cancer. J Urol 181: 872-877.
- Chen HX, Liu YJ, Zhou XD, Luo RY (2005) Expression of cellular FLICE/caspase-8 inhibitory protein is associated with malignant potential in endometrial carcinoma. Int J Gynecol Cancer 15: 663-670.
- Haag C, Stadel D, Zhou S, Bachem MG, Möller P, et al. (2011) Identification of c-FLIPL and c-FLIPS as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut 60: 225-237.
- Schmid SJ, Glatzel MC, Welke C, Kornmann M, Kleger A, et al. (2013) Absence of FLICE- inhibitory protein is a novel independent prognostic marker for very short survival in pancreatic ductal adenocarcinoma. Pancreas 42: 1114-1119.
- Peter ME (2004) The flip side of FLIP. Biochem J 382: 1-3.
- Koyama S (2003) Differential expression of intracellular apoptotic signaling molecules in tumor and tumor-infiltrating lymphocytes during development of invasion and/or metastasis of gastric carcinoma. Dig Dis Sci 48: 2290-2300.
- Ryang DY, Joo YE, Chung KM, Lim SR, Jeong HK, et al. (2007) Expression of c-FLIP in gastric cancer and its relation to tumor cell proliferation and apoptosis. Korean J Intern Med 22: 263-269.
- Tian F, Lu JJ, Wang L, Li L, Yang J, et al. (2012) Expression of c-FLIP in malignant melanoma, and its relationship with the clinicopathological features of the disease. Clin Exp Dermatol 37: 259-265.
- Zhang W, Zhou J, Zhu X, Yuan H (2017) MiR-126 reverses drug resistance to TRAIL through inhibiting the expression of c-FLIP in cervical cancer. Gene 627: 420-427.
- Huang Y, Yang X, Xu T, Kong Q, Zhang Y, et al. (2016) Overcoming resistance to TRAIL- induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int J Oncol 49: 153-163.
- Kim EA, Kim SW, Nam J, Sung EG, Song IH, et al. (2016) Inhibition of c-FLIPL expression by miRNA-708 increases the sensitivity of renal cancer cells to anti-cancer drugs. Oncotarget 7: 31832-31846.
- Carson R, Celtikci B, Fenning C, Javadi A, Crawford N, et al. (2015) HDAC inhibition overcomes acute resistance to MEK inhibition in BRAF-mutant colorectal cancer by downregulation of c-FLIPL. Clin Cancer Res 21: 3230-3240.
- Safa AR (2016) Resistance to cell death and its modulation in cancer stem cells. Crit Rev Oncog 2: 203-219.
- Lee S, Yoon CY, Byun SS, Lee E, Lee SE (2013) The role of c-FLIP in cisplatin resistance of human bladder cancer cells. J Urol 189: 2327-2334.
- Hassanzadeh A, Hagh MF, Alivand MR, Akbari AAM, Asenjan KS, et al. (2018) Down-regulation of intracellular anti-apoptotic proteins, particularly c-FLIP by therapeutic agents; the novel view to overcome resistance to TRAIL. J Cell Physiol 233: 6470-6485.
- Zhitao J, Long L, Jia L, Yunchao B, Anhua W (2015) Temozolomide sensitizes stem-like cells of glioma spheres to TRAIL-induced apoptosis via upregulation of casitas B-lineage lymphoma (c-Cbl) protein. Tumour Biol 36: 9621-9630.
- Piggott L, Omidvar N, Martí-Pérez S, French R, Eberl M, et al. (2011) Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL. Breast Cancer Res 13: 88.
- Hashimoto N, Tsunedomi R, Yoshimura K, Watanabe Y, Hazama S, et al. (2014) Cancer stem-like sphere cells induced from de-differentiated hepatocellular carcinoma-derived cell lines possess the resistance to anti-cancer drugs. BMC Cancer 14: 722.
- Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K (2015) Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis 2: 152-163.
- Thakur B, Ray P (2017) Cisplatin triggers cancer stem cell enrichment in platinum-resistant cells through NF-κB-TNFα-PIK3CA loop. J Exp Clin Cancer Res 36: 164.
- Piggott L, Silva A, Robinson T, Santiago-Gómez A, Simões BM, et al. (2018) Acquired resistance of ER-positive breast cancer to endocrine treatment confers an adaptive sensitivity to TRAIL through posttranslational downregulation of c-FLIP. Clin Cancer Res 24: 2452-2463.
- Eustace AJ, Conlon NT, McDermott MSJ, Browne BC, O'Leary P (2018) Development of acquired resistance to lapatinib may sensitise HER2-positive breast cancer cells to apoptosis induction by obatoclax and TRAIL. BMC Cancer 18: 965.
- Longley DB, Wilson TR, McEwan M, Allen WL, McDermott U, et al. (2006) c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 25: 838-848.
- Klingbeil O, Lesche R, Gelato KA, Haendler B, Lejeune P (2016) Inhibition of BET bromodomain-dependent XIAP and FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic agents. Cell Death Dis 7: e2365.
- Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, et al. (2014) Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 41: 947-959.
- Weiss JM, Subleski JJ, Back T, Chen X, Watkins SK, et al. (2014) Regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment undergo Fas-dependent cell death during IL-2/αCD40 therapy. J Immunol 192: 5821-5829.
- Wesolowski R, Markowitz J, Carson WE (2013) Myeloid derived suppressor cells: A new therapeutic target in the treatment of cancer. J Immunother Cancer 1: 10.
- Kao J, Ko EC, Einstein S, Sikora AG, Fu S, et al. (2011) Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol 77: 12-19.
- Zhao X, Rong L, Zhao X, Li X, Liu X, et al. (2012) TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest 122: 4094-4104.
- Wu YJ, Wu YH, Mo ST, Hsiao HW, He YW, et al. (2015) Cellular FLIP inhibits myeloid cell activation by suppressing selective innate signaling. J Immunol 195: 2612-2623.
- Fossati S, Ghiso J, Rostagno A (2012) TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer's Aβ. Cell Death Dis 3: e321.
- Chiou SH, Chen SJ, Peng CH, Chang YL, Ku HH, et al. (2006) Fluoxetine up-regulates expression of cellular FLICE-inhibitory protein and inhibits LPS-induced apoptosis in hippocampus-derived neural stem cell. Biochem Biophys Res Commun 343: 391-400.
- Huang M, Liang Y, Chen H, Xu B, Chai C, et al. (2018) The role of fluoxetine in activating Wnt/β-catenin signaling and repressing β-amyloid production in an Alzheimer mouse model. Front Aging Neurosci 10: 164.
- Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH (2018) Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: Role of cigarette smoke exposure. Am J Respir Cell Mol Biol 58: 157-169.
- Pouwels SD, Faiz A, Den-Boef LE, Gras R, Berge MVD (2017) Genetic variance is associated with susceptibility for cigarette smoke-induced DAMP release in mice. Am J Physiol Lung Cell Mol Physiol 313: 559-580.
- Wu YH, Kuo WC, Wu YJ, Yang KT, Chen ST, et al. (2014) Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ 21: 451-461.
- Kim HS, Mendiratta S, Kim J, Pecot CV, Larsen JE, et al. ( 2013) Systematic identification of molecular subtype?selective vulnerabilities in non?small?cell lung cancer. Cell 155: 552-566.
- Safa AR. C-FLIP: Encyclopedia of signaling molecules, 2018, 1039-1048. Springer International Publishing AG, Editor, Sangdun Choi.