Keywords
Cholecystokinin; Diabetes Mellitus, Type I; Diabetes Mellitus, Type II; Islets of Langerhans
Abbreviations
CCK: cholecystokinin; CCK- 8: cholecystokinin octapeptide; OGTT: oral glucose tolerance test; OLETF: Otsuka Long- Evans Tokushima fatty; PCNA: proliferating cell nuclear antigen; STZ: streptozotocin
INTRODUCTION
In the adult state, pancreatic beta cells are commonly quiescent. The islets of Langerhans have a very low replicative rate, approximately 2-3% per 24 h in the adult pancreas [1]. The interest in eliciting neogenesis or replication of the endocrine pancreas is considerable because the beta cell mass is largely reduced in diabetes mellitus. Some biological and experimental situations have suggested that the low replicative state of beta cells could be reversed. In rats, during pregnancy, the beta cell mass in islets of Langerhans increased about 50% [2]. Islet neo-formation could be also induced by a 90% partial pancreatectomy completed by treatment with nicotinamide [3]. In fact, small pancreatic ductules could differentiate into new pancreatic islets after a 90% partial pancreatectomy, suggesting the presence of stem cells in the adult pancreas [1].
Among the different growth factors implicated in the dynamics of the beta cell mass and its activity, CCK could play a possible role. CCK-related peptides have been shown to regulate a large number of physiological functions, including not only the secretion of pancreatic enzymes but also the growth and differentiation of the normal and cancerous pancreas [4, 5, 6, 7]. Generally, these effects are mediated by CCK-A receptors present in both ductular and acinar cells [8, 9].
Evidence of CCK-producing cells has been demonstrated in adult rat islets by in situ hybridization and immunoreactivity [10]. In these cells, CCK-8, which is the smallest form capable of retaining a biological activity, has been found to be the major form of CCK [10]. CCK-A receptors have been recognized by immunohistochemistry in insulin and glucagon cells in rat, pig and human pancreata but not in somatostatin cells [11]. These results are consistent with those obtained in genetically diabetic Otsuka Long- Evans Tokushima fatty (OLETF) rats, devoid of CCK-A receptors in the pancreas, and completely insensitive to exogenous and endogenous CCK stimulation, as regards endocrine and exocrine pancreatic secretions [12]. OLETF rats are type 2 or non-insulindependent diabetic rats characterized by hyperglycemia, obesity, hyperinsulinemia and insulin resistance [13]. However, in Zucker rats, another model of type 2 diabetes, the exocrine pancreatic response to CCK was only reduced [14]. This model is characterized by obesity, hyperinsulinemia, increased pancreatic insulin content, insulin resistance and hyperlipidemia [14, 15]. In humans, the effects of CCK on the endocrine pancreas are controversial. According to Kim et al. [16], CCK did not increase serum insulin concentrations in type 2 diabetic subjects in response to intravenous glucose administration, when compared to healthy controls. In contrast, according to Ahren et al. [17], CCK-8 potentiated the increase of circulating insulin and reduced the increase in circulating glucose after meal ingestion, suggesting that CCK exerts an antidiabetogenic effect.
In insulin dependent type 1 diabetic rats, induced by streptozotocin and characterized by hyperglycemia, hypoinsulinemia and a destruction of pancreatic beta cells, low doses of CCK seemed to reduce pancreatic beta cell number and activity [18].
However, in non-diabetic animals, CCK is known to stimulate insulin secretion. In rats, in vivo and in vitro experiments have shown that CCK-8 contributes to the increase of insulin secretion [19]. Similar results have been reported in conscious sheep [20]. In humans, there is disagreement due to different experimental conditions, such as different doses, or the forms of CCK used [21, 22].
Thus, the question remains as to whether CCK-8 possesses the potential abilities of regulating pancreatic beta cell mass and activity in insulin dependent (type 1) and noninsulin dependent (type 2) diabetic rats by stimulating beta cells from a resting state and allowing them to enter into an active cell cycle. Type 1 diabetes was been chemically induced by streptozotocin. The type 2 diabetes model is a new one induced by streptozotocin and partially protected with a suitable dose of nicotinamide [23]. This model, which shares a number of similarities with human type 2 diabetes, is characterized by moderately stable hyperglycemia, glucose intolerance, altered but significant glucosestimulated insulin secretion, responsiveness to tolbutamide and a reduction of pancreatic beta cell mass [23, 24]. Therefore, type 1 and type 2 diabetic rats were treated for 8 successive days with CCK-8 in order to morphometrically investigate the beta-cell population of the islets of Langerhans and to analyze the functional state of these cells by evaluating the glucose response to an oral glucose challenge. The concentrations of CCK-8 chosen were those which induced the most obvious growth effect on the exocrine pancreas in non-diabetic and type 2 diabetic rats (1 and 2 μg/kg respectively, three times daily) and a higher concentration (4 μg/kg, three times daily) which induced a lesser effect or no effect at all in these rats as previously described by us [25].
METHODS
Materials
Sulphated CCK-8 was obtained from Neosystem (Strasbourg, France). The streptozotocin, nicotinamide and hydrolyzed gelatin were from Sigma-Aldrich Chimie (L’Isle d’Abeau Chesnes, France). The glucometer (Prestige) and sticks were provided by Chronolyss (Le Raincy, France). Rabbit polyclonal antibodies to insulin, mouse monoclonal antibodies to PCNA (Proliferating Cell Nuclear Antigen), the streptavidin “LAB” DAB (Diaminobenzidine) kit and chromogens NBT/BCIP (nitro-blue tetrazolium/5-Bromo-4-chloro-3 indolylphosphate) were from SPECI (Varennes-sur-Allier, France).
Animals and Induction of Type 1 and Type 2 Diabetes
One-hundred and 86 male Wistar rats weighing 220-240 g (10 weeks of age) were obtained from DEPRE (St Doulchard, France). They were housed under controlled standard conditions (light/dark cycle, 07:00- 19:00 h lights on), in an ambient temperature of 21±2°C, with food and water available ad libitum.
Seventy-two rats were divided into three groups of 24 animals. Type 2 diabetes was induced in the first group by an intraperitoneal administration of nicotinamide (260 mg/kg body weight) dissolved in saline, 15 minutes before an intravenous injection of streptozotocin (65 mg/kg body weight). Type 1 diabetes was induced in the second group by an intravenous injection of streptozotocin (65 mg/kg body weight) dissolved in 10 mM citrate buffer. The control group was injected with saline buffer.
Fifteen days after the induction of diabetes, the first group exhibited a significant elevation of blood glucose (130-200 mg/dL) without weight loss. The second group showed growth retardation, polyuria, polydipsia and a marked elevation of blood glucose (higher than 350 mg/dL).
Then, the animals in each group were subdivided into 4 subgroups of 6 animals each. Each subgroup received either saline (the control group) or CCK-8 injections at various concentrations: 1, 2 and 4 μg/kg body weight for 8 successive days. Subcutaneous injections were carried out three times daily, the CCK-8 being added to 15% hydrolyzed gelatin. During the entire experiment, the animals were weighed every day and fed ad libitum with a chow diet (UAR, Villemoissonsur- Orge, France).
On day 9, the animals were sacrificed by exsanguination. The pancreata were quickly removed, freed from fat and connective tissue, and processed for immunohistochemistry.
Another 72 rats were subdivided into 3 additional groups of 24 rats each (nondiabetic rats, type 1 and type 2 diabetic rats) and were treated as described above. The animals were treated for 8 successive days with saline (controls) or CCK-8 at different concentrations (1, 2 and 4 μg/kg). On day 9, they were sacrificed after a four-hour fast. Blood was collected for glucose and insulin analysis. The pancreata were quickly removed, freed from fat and connective tissue, weighed and stored at -20°C in order to carry out pancreatic insulin assays.
Immunohistochemistry
Pancreatic fragments were fixed by immersion in 10% buffered formalin, dehydrated and embedded in paraffin. Five micrometer sections were cut and doublestained for Proliferating Cell Nuclear Antigen (PCNA) and for beta-cell hormone insulin. In brief, after removal of the paraffin, the slices were incubated for 30 min at room temperature with the primary antibody directed against PCNA (dilution 1:50). The slices were rinsed and incubated with the secondary antibody marked with biotin. Then, they were rinsed again and incubated in streptavidin-peroxydase complex for 25 min. Finally, they were revealed with DAB (diaminobenzidine) solution for 1 min. Thereafter, the slices underwent insulin staining. The sections were incubated for 30 min in a buffer containing polyclonal antiinsulin antibody diluted at 1:100. After rinsing, the sections were incubated for 30 min with anti-rabbit anti-insulin antibody coupled with alkaline phosphatase. The alkaline phosphatase activity was revealed by adding NBT/BCIP (nitro blue tetrazolium/5- bromo-chloro-3-indolyl phosphate) for 5-7 min. The sections were counterstained with hematoxylin. For non-specific staining, the sections were incubated by using the second antibody without the primary antibody, and the specificity was determined by neutralization with excess antigens.
Each section then underwent morphometry using computerized image analysis software (Visioscan 2000, Biocom, Les Ulis, France). The parameters measured included the entire surface of the pancreatic tissue and the surface of each endocrine islet in the pancreatic tissue. In addition, to calculate the mitotic index, PCNA-positive beta-cell nuclei as well as PCNA-negative beta-cell nuclei were counted at a magnification of x630. PCNA-positive beta-cell nuclei were expressed as a percentage of the total PCNAnegative and positive beta-cell nuclei. In nondiabetic rats and in type 2 diabetic rats, a minimum of 2,000 nuclei were scored. In the type 1, at least 1,000 nuclei were scored.
Plasma and Pancreatic Insulin Analyses
Plasma (1,000 μL) was separated by centrifugation (10 min at 1,000 g at 4°C) from the blood samples collected after exsanguination and frozen at -20°C until insulin measurement was carried out. The insulin concentration in plasma was measured directly by the radioimmunological method (Insulin-CT kit from CIS Bio International, Gif-sur-Yvette, France).
For extraction of pancreatic insulin content, the pancreata were homogenized in 5 mL of a cold mixture of HCl:ethanol (1:3, vol/vol) using a polytron and samples were stored 48 h at 4°C. After centrifugation (50 min at 1,000 g at 4°C) and separation of the supernatants, the pellets were extracted again using acidified ethanol for 24 h at 4°C. Then, the homogenates were centrifuged again, and the supernatants obtained after centrifugation were pooled with the previous ones and kept at -20°C until assayed. The pancreatic insulin contents were measured by radioimmunoassay using the same kit as was used for plasma insulin.
CCK Effectiveness In Vivo
Three additional groups of non-diabetic, type 1 and type 2 diabetic rats (n=14 rats each) were generated as described above to perform oral glucose tolerance tests (OGTTs) after CCK-8 administration. Each group was subdivided into two subgroups of 7 rats each. They were treated for 8 successive days either with saline (for controls) or with the most effective CCK-8 concentrations (4 μg/kg for type 1 diabetic rats and 1 μg/kg for nondiabetic and type 2 diabetic rats). Then, OGTTs were performed on the rats after an overnight fast and fasting was continued during the experiment. D-glucose (2 g/kg body weight) was administered endogastrically through a “metallic needle” in conscious rats. Blood samples were collected from the tail vein before and 10, 20, 30, 60, 90 and 120 min after gavage with glucose. Glucose concentrations in the blood were measured by a glucometer (Prestige, Chronolyss, Le Raincy, France).
ETHICS
All animals received human care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals”. Experiments were carried out in accordance with the European Community Council Directive of November 24, 1986 (86/609/EEC).
STATISTICS
In order to evaluate CCK-8 effectiveness in vivo, the areas under the curves (AUC) were calculated using Micropharm software (Loginserm, Paris, France).
All results are represented as mean±SEM. Comparisons among the 3 groups were carried out by means of one-way ANOVA followed by a multiple comparison post-test (Bonferroni) while comparisons between CCK-8-treated and saline-treated rats were evaluated by means of one-way ANOVA without Bonferroni correction. Two-tailed P values of less than 0.05 were considered statistically significant. All statistical evaluations were performed by running the Instat 2.00 MacInstosh statistical package (Graph Pad Software, San Diego, CA).
RESULTS
Blood Glucose Concentration
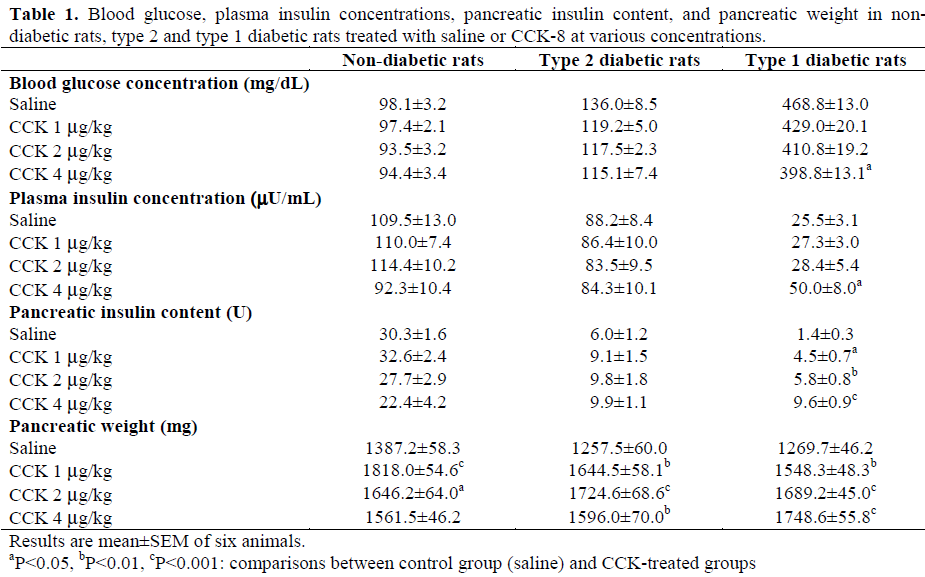
As represented in Table 1, non-diabetic rats showed a blood glucose concentration of 98.1±3.2 mg/dL after 8 days of treatment with saline. In streptozotocin-nicotinamide-treated rats (type 2 diabetes), the blood glucose concentration was higher than that in nondiabetic rats (136.0±8.5 mg/dL, P<0.015). In streptozotocin-treated rats (type 1 diabetes), the blood glucose concentration was markedly increased when compared to non-diabetic rats (468.8±13.0 mg/dL, P<0.001). After eight days of CCK-8 treatment in type 1 diabetic rats, the blood glucose concentration decreased in a dose-dependent manner; the maximum effect was -15% (P<0.014 vs. saline) being observed with 4 μg/kg CCK-8. However, in non-diabetic rats, blood glucose concentration was not affected by CCK treatment while in type 2 diabetics, there was a slight but not significant reduction of this parameter (Table 1).
Plasma Insulin and Pancreatic Insulin Analyses
After eight days of saline treatment, the plasma insulin concentration in non-diabetic rats was 109.5±13.0 μU/mL (Table 1). In type 2 diabetic rats, it was slightly but not significantly lower (88.2±8.4 μU/mL) but in type 1 diabetic rats, it decreased strongly by comparison to non-diabetic rats (25.5±3.1 μU/mL, P<0.001). Long-term administration of CCK-8 did not significantly affect this parameter in non-diabetic rats and type 2 diabetic rats but in type 1 diabetic rats, the plasma insulin concentration increased significantly with the highest concentration of CCK-8 (4 μg/kg vs. saline: +96%, P=0.008) (Table 1).
By comparison to non-diabetic rats, the pancreatic insulin concentration was reduced by 80% (P<0.001) in type 2 diabetic rats and by 95% (P<0.001) in type 1 diabetics (Table 1). CCK-8 treatment did not significantly affect pancreatic insulin content in nondiabetic and type 2 diabetic rats though this parameter was slightly increased in this latter group. In contrast, in type 1 diabetic rats, the pancreatic insulin content increased progressively, in a dose dependent manner, with a maximum increase of 586% at the highest concentration of CCK-8 (4 μg/kg vs. saline: P<0.001) (Table 1).
Effects of CCK-8 on Pancreatic Growth
As illustrated in Table 1, CCK-8 administered for 8 successive days in non-diabetic rats, exerted a biphasic effect on pancreatic growth as a function of the concentration used. It increased pancreatic weight, with a maximum effect of 31% at 1 μg/kg concentration (P<0.001 vs. saline), this effect being less obvious with higher concentrations of CCK-8. Indeed, this increase was less pronounced (12.6%, P=0.052 vs. saline) at the concentration of 4 μg/kg. In type 2 diabetic rats, the profile of pancreatic weight appeared to be similar but the maximum increase (37%) was observed at the CCK-8 concentration of 2 μg/kg (P<0.001). In type 1 diabetic rats, the dose-response curve for pancreatic weight to CCK-8 was shifted toward the higher CCK-8 dose. Indeed, the maximum increase in pancreatic weight (38%) was noted with the CCK-8 concentration of 4 μg/kg (P<0.001 vs. saline).
Thus, CCK-8 treatment stimulated pancreatic growth in the rat with a maximum effect at 1μg/kg, 2 μg/kg and 4 μg/kg for non-diabetic rats, type 2 diabetic rats and type 1 diabetic rats, respectively.
Histological Analysis
In non-diabetic rats, the pancreas showed numerous round shaped pancreatic islets of different sizes. The islets were randomly scattered throughout the acinar tissue. Pancreatic islets are mainly composed of beta cells, characterized by their staining for insulin in blue (Figure 1A). These cells are localized in the central part of the islets. Replicative beta cells are double stained with insulin in the cytoplasm in blue and with PCNA in the nucleus in brown. The CCK-8 treatment had no effect on the morphological aspect of the islets in non-diabetic rats.
Figure 1. Immunohistochemical analysis of PCNA and
insulin in pancreatic tissue from non-diabetic rats (A),
type 2 diabetic rats (B) and type 1 diabetic rats (C).
Arrows indicate PCNA-stained beta cells in islets.
Extra-islet beta cells localized in pancreatic ducts from
type 2 diabetic rats (D). Magnification x630.
In type 2 diabetic rats, the pattern of islet cell distribution and the morphology of the islets were similar to those of non-diabetic rats (Figure 1B). The pancreatic sections from type 2 diabetic rats after a 4 μg/kg CCK-8 administration showed extra-islet beta cells closely related to the ductular epithelium (Figure 1D). There were fewer extra-islet beta cells in the non-diabetic groups and were not observed in type 1 diabetic rats.
In contrast to the two other groups, the pancreata from type 1 diabetic rats exhibited a severe degeneration of the pancreatic islets due to streptozotocin injection. The pancreatic islets were reduced in number, size and in insulin-immunoreactive cells (Figure 1C). After CCK-8 treatment, the beta cells did not show an improved morphology but PCNAlabeling was more obvious in these cells.
In the exocrine pancreas, abundant PCNA labeling was observed on both acinar and ductular cells from the three experimental groups treated with CCK-8. However, we never observed any sign of pancreatitis even at the highest concentration of CCK-8.
Morphometrical Analysis
The morphometrical analysis of the surface of endocrine islets revealed that diabetes decreased the surface of the islets of Langerhans by 23% (P=0.247) in type 2 diabetic rats and by 61% (P<0.008) in type 1 diabetic rats when compared to non-diabetic control rats (Table 2). CCK-8 treatment did not significantly alter this parameter either in non-diabetic rats or in type 2 diabetic rats but significantly increased it in type 1 diabetic rats; the maximum increase (50%) being noted with 4 μg/kg CCK-8 (P<0.011 vs. saline) (Table 2).
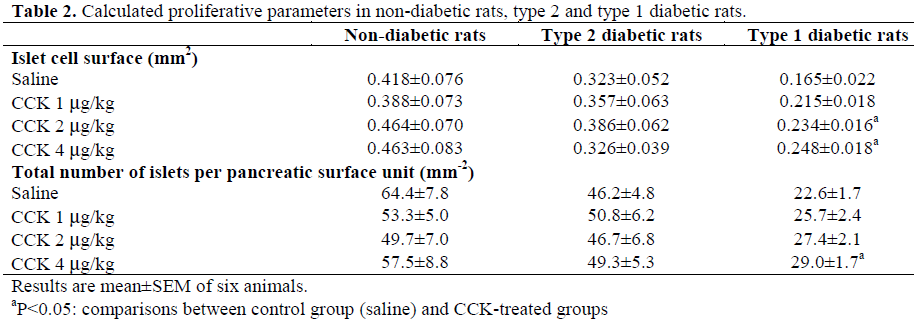
The number of islets decreased by 28% (P=0.037) and 65% (P<0.001) in type 2 and type 1 diabetic rats respectively when compared to non-diabetics (Table 2). Chronic treatment with CCK-8 did not modify this parameter in non-diabetic and type 2 diabetic rats but increased it (28%) in type 1 diabetic rats at a concentration of 4 μg/kg CCK-8 (P=0.023) (Table 2).
The mitotic index characterized by the percentage of PCNA-stained beta cells to the total number of beta cells in the three experimental groups is illustrated in Figure 2. By comparison to non-diabetic control animals, this parameter decreased by 36% (P=0.071) in type 2 diabetic rats and by 73% (P<0.001) in type 1 diabetic rats. CCK-8 treatment did not significantly alter the mitotic index in non-diabetic animals as well as in type 2 diabetic rats. In contrast, in type 1 diabetic rats, CCK-8 treatment significantly increased (P<0.001) this parameter by 280%, 325%, and 380% with the respective concentrations of 1, 2 and 4 μg/kg (Figure 2). Thus, CCK-8 stimulates the proliferation of beta cells in type 1 diabetic animals but not in non-diabetic rats or in type 2 diabetic rats.
Figure 2. Beta cell proliferation in islets from nondiabetic
rats (dotted columns), type 2 diabetic rats
(hatched columns) and type 1 diabetic rats (black
columns) treated with saline or CCK-8 (1, 2 and 4
μg/kg). Pancreata slices were immunostained for
PCNA and insulin. After counterstaining, at least 2000
nuclei in non-diabetic rats and type 2 diabetic rats or
1000 nuclei in type 1 diabetic rats were counted per
pancreas. The data (mean±SEM) are represented as
percentages of PCNA labeled beta cells to the total
number of insulin labeled beta cells.
**P<0.001; comparisons between CCK-8 treated (n=6)
and saline treated (n=6) rats
CCK-8 Effectiveness In Vivo
The oral glucose tolerance test was performed in rats undergoing an overnight fast. As shown in Figure 3, after a peroral glucose administration (2 g/kg of body weight), blood glucose concentrations increased up to 20 minutes in both non-diabetic and type 2 diabetic rats and up to 30 min in type 1 diabetic rats. Then the blood glucose concentration decreased again and returned progressively to the control values observed before the peroral administration of glucose in both former groups but not in the latter group. However, the maximum increase in blood glucose concentration was 86% of basal values (P<0.001) in non-diabetic controls, 188% (P<0.001) in type 2 diabetic rats and 198% (P<0.001) in type 1 diabetic rats. Thus, after gavage with glucose, blood glucose concentrations attained significantly higher values in type 1 diabetic rats than in type 2 diabetics (381±36 mg/dL after 30 min vs. 188±15 mg/dL after 20 min; P<0.001), the lowest responses being observed in nondiabetic controls (146±6 mg/dL after 20 min). CCK-8 treatment at the concentration of 1 μg/kg in non-diabetic rats and in type 2 diabetic rats did not significantly change this profile. Indeed, the maximum increases in blood glucose concentration in CCK-8 treated rats, observed at the same periods (20 min), CCK-8 treatment did not improve the functional state of beta cells in the three groups of animals.
Figure 3. Blood glucose concentrations during OGTT
in non-diabetic rats (A), type 2 diabetic rats (B) and
type 1 diabetic rats (C) treated with saline (open
squares) or CCK-8 (1 μg/kg for non-diabetic and type
2 diabetic rats or 4 μg/kg for type 1 diabetic rats)
(black squares).
Insets: AUC for the glycemic responses (mg/dL x min
over a 120-min test) in saline (controls, white columns)
and CCK-8 treated rats (black columns). Glucose was
administered at a dosage of 2 g/kg. Data are
mean±SEM of 7 animals.
CCK-8 treatment did not improve the functional state of beta cells in the three groups of animals.
DISCUSSION
In the present study, we examined the effect of CCK-8 chronic treatment on type 1 and type 2 diabetic animal models to induce pancreatic islets cell proliferation and differentiation.
We demonstrated that the 8-day-treatment leads to the formation of new islet beta cells in type 1 diabetic rats. This effect is dosedependent, the more obvious effect being noted at a concentration of 4 μg/kg CCK-8. Beta cells are able to improve basal glucose and insulin concentrations but are not sufficient to significantly improve the glycemic response to an oral glucose challenge. However, CCK-8 treatment did not significantly affect the proliferation of betacells in the islets of type 2 diabetic rats and non-diabetic rats.
In the present study, we also found that CCK increased pancreatic weight in non-diabetic rats. This trophic effect, already reported by us [7, 25], can be explained by the proliferation of both acinar and ductular cells markedly labeled in this study with PCNA. These data are in agreement with our previous data, which demonstrated the presence of mitotic figures in pancreatic cells after a 4- day-treatment with cerulein, a CCK-analogue [6]. However, in non-diabetic rats, the optimal growth response of CCK-8 was induced by the lowest concentration of the peptide, i.e. 1 μg/kg. These results are consistent with our previous data which in addition, showed the presence of two classes of CCK-8 binding sites (with high and low affinity) in nondiabetic rats [25]. In type 2 diabetic rats, CCK-8 also increased pancreatic weight, but the maximum effect was noted at 2 μg/kg CCK-8 as described previously [25]. In type 1 diabetic rats, CCK-8 had similar effects on the exocrine pancreas but, in this case, higher CCK-8 concentrations were necessary to produce this effect. Thus, the maximum growth effect was noted at 4 μg/kg CCK-8, suggesting a decreased sensitivity to CCK-8. In the type 2 diabetes model, the loss of sensitivity to CCK-8 was attributed to the loss of CCK1 receptors of high affinity [25]. A similar mechanism should be hypothesized for the type 1 diabetes model. Nevertheless, the intense PCNA-labeling in both acinar and ductular cells from type 1 and type 2 diabetic rats suggests that pancreatic growth is due to the proliferation of both types of cells.
According to our morphometric analysis of pancreatic islets from non-diabetic rats, neither the islet surface, the number of islets or the beta cell proliferation were affected by CCK-8 chronic administration. Moreover, CCK-8 failed to improve glucose-induced insulin secretion or pancreatic insulin content. Thus, in contrast to the exocrine pancreas, long-term CCK-8 treatment did not affect the structure and function of pancreatic islets in normoglycemic rats.
Our type 2 diabetes model, induced by the simultaneous administration of nicotinamide and streptozotocin, is characterized by the decreased size and number of islets of Langerhans associated with a reduction of pancreatic insulin content, a moderate increase in basal blood glucose concentration and an impaired glucose response to an oral glucose challenge. This profile correlates with the description of the same model proposed by Masiello et al. [23]. These authors also reported limited concentrations of circulating insulin after glucose administration in rats [23]. Thus, the nicotinamide/streptozotocin induced type 2 diabetic model in rats is characterized by abnormalities in glucose tolerance and insulin responsiveness, alterations also encountered in type 2 diabetes in humans.
In the experimental model for type 2 diabetes, chronic administration of CCK-8 was not able to increase islet cell surface and number. In addition, CCK-8 treatment did not significantly alter plasma insulin concentration but tended to slightly increase pancreatic insulin content. This effect may be correlated to an increase in the number of extra-islet beta cells which were not included in the total islet cell surface. Indeed, our histological observation showed endocrine cells generated by the ductular epithelium in the type 2 diabetic animals receiving CCK-8 treatment. These new cells could represent islet neogenesis which has been welldescribed in different models of islet regeneration such as pancreatic duct ligation, partial pancreatectomy or cellophane wrapping of the pancreas [1, 26, 27, 28, 29]. But, this neoformation was not sufficient to influence either the basal blood glucose concentrations or those after oral glucose administration. Thus, the restoration of insulin content to higher concentrations by CCK-8 failed to decrease the hyperglycemic peak after OGTT assays. Our results agree with those of Rushakoff et al. and Schmid et al. who demonstrated that CCK-8 had no effect on glucose-stimulated insulin secretion in humans [30, 31]. In contrast, Ahren et al. reported that CCK-8 could elicit a decrease in plasma glucose concentrations in patients after meal ingestion [17]. In this case, CCK-8 exerted a direct stimulation on insulin secretion, playing an antidiabetogenic role similar to that played by GLP-1 for type 2 diabetes.
Our type 1 diabetic model induced by streptozotocin is characterized by very high concentrations of basal blood glucose concentration, associated with a markedly impaired glycemic response to an oral glucose challenge, a strong reduction of pancreatic insulin content, a decrease of plasma insulin concentration and, from the histological point of view, an obvious reduction of the size and number of islets of Langerhans in which beta cells are less proliferative. These alterations have also been reported by others [18]. In this experimental model for type 1 diabetes, longterm CCK-8 treatment induced the proliferation of beta cells as shown by a marked increase of the mitotic indices correlated with the increase in the concentration of the peptide administered. Indeed, the ratio of PCNA labeled beta cells to the total number of insulin-labeled cells increased by 380% with the highest concentration of CCK-8, i.e. 4 μg/kg body weight. These results are in disagreement with those of Takacs et al. [18] who reported that CCK-8 failed to promote pancreatic regeneration in streptozotocin-induced diabetic rats following the induction of experimental pancreatitis with arginine. But the concentration of CCK-8 used by these authors was very low, 1 μg/kg given twice daily while we gave the same concentration as well as higher doses (2 and 4 μg/kg) three times daily. It is clear that at the lowest concentration, the modifications of the different parameters we analyzed were not the most evident. By comparison to our type 2 diabetes model, in which the hyperglycemic state is limited and CCK-8 did not stimulate beta cell proliferation in the islets, it seems that the marked hyperglycemic state in our type 1 diabetic rats could potentiate the action of CCK-8 on the proliferation of beta cells. These results agree with those of Zawalich et al. [32] who demonstrated that CCK-8 stimulated insulin secretion from isolated rat pancreatic islets only when high glucose concentrations were present. Hardikar et al. [33] also reported that pre-exposure of fetal porcine beta-cells to CCK increased insulin secretion in the presence of high glucose concentrations but not in low concentrations, and enhanced the formation of beta-cells from undifferentiated precursors. Thus, glucose sensitizes the islets to the action of CCK-8. In fact, glucose by itself has been shown to be a mitotic factor for beta cells [34]. Thus, according to our results, CCK-8 seems to be able to stimulate beta cells from the resting state and allow them to enter into an active cell cycle only in the presence of high glucose concentrations. Our morphometrical analysis indicates that these modifications are accompanied by an increase in both islet cell surface and number. In consequence, plasma insulin concentrations increased in streptozotocin-induced diabetic rats treated with CCK-8 while basal glucose concentrations decreased. However, the glycemic response to a glucose tolerance test was not significantly improved, indicating that these newly formed beta cells were not mature enough to induce full beta cell functionality. Perhaps, longer CCK-8 treatment is necessary to attain this objective. In summary, the effects of CCK-8 on the proliferation of beta cells are complex. From the comparison of the two models of diabetes - one with a limited increase of blood glucose concentration and the other one with a higher glucose concentration - it generally appears that CCK-8 has the capacity of stimulating the proliferation of beta cells but requires the presence of high concentrations of plasma glucose. Further studies are necessary to clarify the true mechanism of action of CCK at the cellular and subcellular level.
Acknowledgements
We thank S Heffner for the expert technical assistance and Chronolyss (Le Raincy, France) for providing the glucometers and sticks
References
- Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 1993; 42:1715-20. [PMID 8243817]
- Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992; 130:1459-66. [PMID 1537300]
- Yonemura Y, Takashima T, Miwa K, Miyazaki I, Yamamoto H, Okamoto H. Amelioration of diabetes mellitus in partially depancreatized rats by poly(ADPribose) synthetase inhibitors. Evidence of islet B-cell regeneration. Diabetes 1984; 33:401-4. [PMID 6323238]
- Logsdon CD. Stimulation of pancreatic acinar cell growth by CCK, epidermal growth factor, and insulin in vitro. Am J Physiol 1986; 251:G487-94. [PMID 3020992]
- Axelson J, Hakanson R, Ihse I, Lilja I, Rehfeld JF, Sundler F. Effects of endogenous and exogenous cholecystokinin and of infusion with the cholecystokinin antagonist L-364,718 on pancreatic and gastrointestinal growth. Scand J Gastroenterol 1990; 25:471-80. [PMID 2359975]
- Damgé C, Hajri A, Lhoste E, Aprahamian M. Comparative effect of chronic bombesin, gastrinreleasing peptide and caerulein on the rat pancreas. Regul Pept 1988; 20:141-50. [PMID 2452459]
- Hajri A, Damgé C. Effects of cholecystokinin octapeptide on pancreatic acinar carcinoma in the rat. Pharm Res 1998; 15:1767-74. [PMID 9834001]
- Hajri A, Aprahamian M, Damgé C. Effect of a new CCK-receptor antagonist, CR 1409, on pancreatic growth induced by caerulein, CCK-8, bombesin and gastrin-releasing peptide in the rat. Digestion 1989; 43:66-72. [PMID 2806757]
- Povoski SP, Zhou W, Longnecker DS, Jensen RT, Mantey SA, Bell RH Jr. Stimulation of in vivo pancreatic growth in the rat is mediated specifically by the way of cholecystokinin-A receptors. Gastroenterology 1994; 107:1135-46. [PMID 7523219]
- Shimizu K, Kato Y, Shiratori K, Ding Y, Song Y, Furlanetto R, et al. Evidence for the existence of CCKproducing cells in rat pancreatic islets. Endocrinology 1998; 139:389-96. [PMID 9421438]
- Morisset J, Julien S, Lainé J. Localization of cholecystokinin receptor subtypes in the endocine pancreas. J Histochem Cytochem 2003; 51:1501-13. [PMID 14566022]
- Tachibana I, Akiyama T, Kanagawa K, Shiohara H, Furumi K, Watanabe N, Otsuki. M. Defect in pancreatic exocrine and endocrine response to CCK in genetically diabetic OLETF rats. Am J Physiol 1996; 270:730-7. [PMID 8928805]
- Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes 1992; 41:1422-8. [PMID 1397718]
- Stern JS, Johnson PR, Batchelor BR, Zucker LM, Hirsch J. Pancreatic insulin release and peripheral tissue resistance in Zucker obese rats fed high- and low-carbohydrate diets. Am J Physiol 1975; 228:543-8. [PMID 1091158]
- Zucker LM. Hereditary obesity in the rat associated with hyperlipemia. Ann N Y Acad Sci 1965; 131:447-58. [PMID 5216982]
- Kim KH, Lee HS, Kim CD, Chun HJ, Song CW, Um SH, et al. Evaluation of pancreatic exocrine function using pure pancreatic juice in non insulindependent diabetes mellitus. J Clin Gastroenterol 2000; 31:51-4. [PMID 10914777]
- Ahren B, Pettersson M, Uvnas-Moberg K, Gutniak M, Efendic S. Effects of cholecystokinin (CCK)-8, CCK-33, and gastric inhibitory polypeptide on basal and meal-stimulated pancreatic hormone secretion in man. Diabetes Res Clin Pract 1991; 13:153-162. [PMID 1683622]
- Takacs T, Hegyi P, Jarmay K, Czako L, Gog C, Rakonczay Z Jr, et al. Cholecystokinin fails to promote pancreatic regeneration in diabetic rats following the induction of experimental pancreatitis. Pharmacol Res 2001; 44:363-72. [PMID 11712866]
- Zawalich WS, Diaz VA, Zawalich KC. Stimulatory effects of cholecystokinin on isolated perifused islets inhibited by potent and specific antagonist L 364718. Diabetes 1988; 37:1432-7. [PMID 3046973]
- Mineo H, Iwaki N, Kogishi K, Zabielski R, Onaga T, Kato S. Effects of intravenous infusions of cholecystokinin (CCK)-8 on exocrine and endocrine pancreatic secretion in conscious sheep. Comp Biochem Physiol A Physiol 1995; 111:133-8. [PMID 7537612]
- Ahren B, Holst JJ, Efendic S. Antidiabetogenic action of cholecystokinin-8 in type 2 diabetes. J Clin Endocrinol Metab 2000; 85:1043-8. [PMID 10720037]
- Reimers J, Nauck M, Creutzfeldt W, Strietzel J, Ebert R, Cantor P, Hoffmann G. Lack of insulinotropic effect of endogenous and exogenous cholecystokinin in man. Diabetologia 1988; 31:271-80. [PMID 3294066]
- Masiello P, Broca C, Gross R, Roye M, Manteghetti M; Hillaire-Buys D, et al. Experimental NIDDM: development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes 1998; 47:224-9. [PMID 9519717]
- Novelli M, Fabregat ME, Fernandez-Alvarez J, Gomis R, Masiello P. Metabolic and functional studies on isolated islets in a new rat model of type 2 diabetes. Mol Cell Endocrinol 2001; 175:57-66. [PMID 11325516]
- Kuntz E, Pinget M, Damgé C. Effects of cholecystokinin octapeptide on the exocrine pancreas in a new rat model of type 2 diabetes. Eur J Pharmacol 2002; 448:253-61. [PMID 12144949]
- Wang RN, Kloppel G, Bouwens L. Duct- to isletcell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 1995; 38:1405-11. [PMID 8786013]
- Rosenberg L, Brown RA, Duguid WP. A new approach to the induction of duct epithelial hyperplasia and nesidioblastosis by cellophane wrapping of the hamster pancreas. J Surg Res 1983; 35:63-72. [PMID 6865394]
- Rosenberg L, Duguid WP, Vinik AI. The effect of cellophane wrapping of the pancreas in the Syrian golden hamster: autoradiographic observations. Pancreas 1989; 4:31-7. [PMID 2654928]
- Hardikar AA. Generating new pancreas from old. Trends Endocrinol Metab 2004; 15:198-203. [PMID 15223048]
- Rushakoff RJ, Goldfine ID, Carter JD, Liddle RA. Physiological concentrations of cholecystokinin stimulate amino acid- induced insulin release in humans. J Clin Endocrinol Metab 1987; 65:395-401. [PMID 3305550]
- Schmid R, Schusdziarra V, Schulte-Frohlinde E, Maier V, Classen M. Effect of CCK on insulin, glucagon, and pancreatic polypeptide levels in humans. Pancreas 1989; 4:653-61. [PMID 2682604]
- Zawalich W, Takuwa N, Takuwa Y, Diaz VA, Rasmussen H. Interactions of cholecystokinin and glucose in rat pancreatic islets. Diabetes 1987; 36:426- 33. [PMID 3028890]
- Hardikar AA, Wang XY, Williams LJ, Kwok J, Wong R, Yao M, Tuch BE. Functional maturation of fetal porcine beta-cells by glucagon-like peptide 1 and cholecystokinin. Endocrinology 2002; 143:3505-14. [PMID 12193564]
- Swenne I. The role of glucose in the in vitro regulation of cell cycle kinetics and proliferation of fetal pancreatic B-cells. Diabetes 1982; 31:754-60. [PMID 6761212]




