Keywords
Citric acid; o-Toluidines; Ethyl chloroacetate; Regioselectivity; N-Alkylated Indazoles
Introduction
Heterocyclic ring systems are frequently found in numerous naturally occurring compounds and they compose the core structures of many biologically active motifs as well as some industrial compounds [1]. Among them, indazoles represent an important class of nitrogen containing heterocycles and this nucleus (Figure 1) [2,3] is of great current interest as partial structure in many synthetic drugs or drug candidates with a broad range of pharmacological activities including anti-HIV [4], anti-inflammatory [5], anti-tumor [6,7], anti-depressant [8], analgesic and antipyretic [9], anti-leukemia [10], anti-convulsant activity [11], anti-cancer [12], anti-arthritic [13], anthelmintic [14] and anti-diabetes [15]. Indazole moiety is present only in three natural products such as Nigellicine, Nigeglanine and Nigellidine indicates their rare presence in nature [16,17].
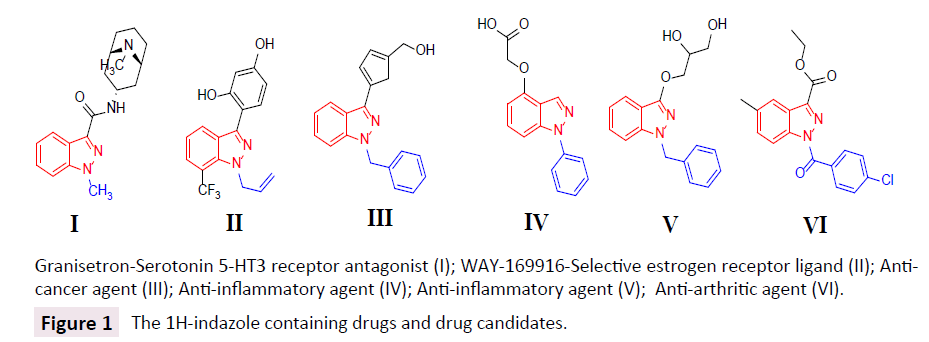
Figure 1: The 1H-indazole containing drugs and drug candidates.
Extensive literature survey revealed that a huge number of methods have been reported for the synthesis of substituted 1H-indazoles. One general route involves diazotization of o-alkylsubstituted anilines followed by acid promoted cyclization [18,19], functionalized sydnone, oxadiazole condensation/Boulton- Katritzky rearrangement [20], intramolecular dehydration from common arylamino oximes/ o-aminobenzoximes [21,22], condensation of arynes with nitrile imines/ diazomethane derivatives/diazo compounds/hydrazones [23,24], condensation of hydrazine to o-halo/o-alkoxy arylhydrazone [25,26]. In addition to the above methods, other intra molecular cyclizations are also reported for the synthesis of indazole derivatives, for example Pd-mediated ring closure of hydrazine and hydrazone based derivatives [27,28], copper catalyzed intramolecular amination of o-haloarylcarbonyl compounds/2-halobenzohydrazides [29,30], CuI promoted cyclization of an aryl hydrazone under microwave irradiation [31], FeBr3/O2-mediated intramolecular C-H amination of arylhydrazones [32], PIFA-mediated oxidative cyclization of o-amidobenzanilides [33], Rhodium-catalyzed cyclocarbonylation of azobenzenes [34], Zinc catalyzed reductive cyclization of o-nitrobenzanilides [35], layered Zirconium sulfophenyl phosphonate catalyzed synthesis from condensation of 1,3-diones and hydrazines under solvent free conditions [36] have been reported.
However, all the above methods are highly promising, but, most of them suffer from one or more disadvantages including the use of i) acids or reagents that are not environmentally compatible and corrosive, ii) hazardous starting materials, iii) metal catalysts and solvent(s), iv) long reaction time, v) cumbersome workup procedures and vi) production of a large amount of waste. In view of the importance of indazoles and the limitations of previous methods, the development of one-pot synthesis of indazoles from readily available starting materials in green catalytic medium would be of prime synthetic value. On the path of finding suitable one-pot green approach, our attention is directed towards classical diazotization and intramolecular cyclization in the presence of AcOH [37]. Prompted by this idea, biodegradable citric acid medium has been identified for the synthesis of N-alkylated 1H-indazoles in a single step operation. Citric acid is the non-toxic, low-cost, biodegradable and environmentally benign catalyst which is also employed in many organic transformations [38-41].
In this work, we demonstrated a biodegradable citric acid mediated green synthetic route for the regioselective synthesis of N-alkylated indazoles in good to excellent isolated yields ( ~ 78- 96%) from readily available starting materials 2-methylanilines (1), NaNO2 (2) and ethyl chloroacetate (3) via diazotization, intra molecular cyclization and followed by N-alkylation in the presence of 1:1 ratio of ethanol and water in a single step operation. If the substrate (1) contains – NO2 group and is reduced to – NH2 in the presence of Fe, CaCl2 at 60-65°C (Chandrappa, Vinaya, Ramakrishnappa, & Rangappa, 2010) in the same step as shown in Figure 2.
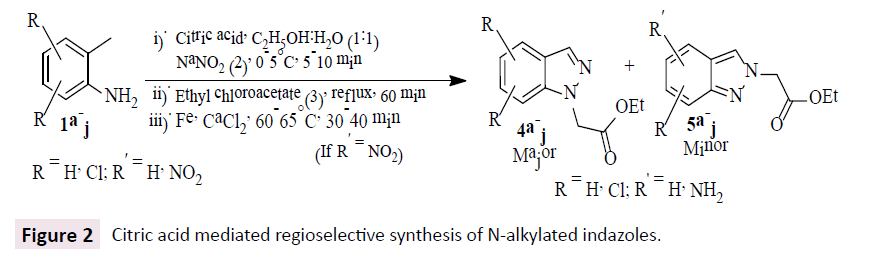
Figure 2: Citric acid mediated regioselective synthesis of N-alkylated indazoles.
Materials and Methods
1H NMR spectra are recorded on a Varian 300 MHz. Chemical shifts are expressed in parts per million (ppm), coupling constants are expressed in Hertz (Hz). Splitting patterns describe apparent multiplicities and are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Mass spectra (MS) are acquired on a Waters LCT premier XE TOF with electron spray ionization (ESI) source. Thin-layer chromatography is performed on 0.25 mm Merck silica gel plates and visualized with UV light. Column chromatography is performed on silica gel. Chemicals are purchased from Sigma Aldrich and Acros organics. Solvents are purchased from Merck. Millipore deionized water is used. Isolated compounds are identified on the basis of spectroscopic data and Mass spectrometry.
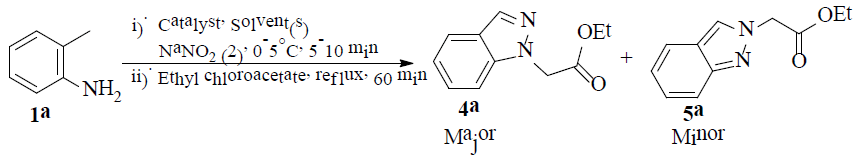
General experimental procedure for (substrates without – NO2 functional group) the regioselective synthesis of ethyl 2-(1H-indazol-1-yl)acetate (4a): In a 100 mL three necked round bottom flask, 2-methylaniline (1a) (10 mmol) and citric acid (20 mmol) are dissolved in a mixture of ethanol (8 mL) and water (4 mL). The clear solution was cooled at 0-5°C and stirred well. To this added ice-cold solution of NaNO2 (2) (12 mmol) in 4 mL of water in 4 portions for 5-10 minutes. The formation of 1H-indazole was monitored by TLC. After the completion of the reaction as per TLC, ethyl chloroacetate (3) (12 mmol) is added to the reaction mixture and is stirred under reflux conditions for 60 min. The progress of the reaction is monitored by TLC. After completion of the reaction, the reaction mixture is quenched in ice and the crude product is extracted twice with ethyl acetate (2 × 10 mL). The organic layer is washed with brine, dried over MgSO4 and concentrated. The obtained crude product (4a) is purified by silica gel using n-hexane and EtOAc (3:1 ratio). The same procedure is followed for the preparation of compounds 4b-e. The synthesized compounds (4a-e) gave satisfactory spectroscopic data in accordance with their proposed structures.
General experimental procedure for the (substrates with – NO2 functional group) regioselective synthesis of ethyl 2-(5-amino-1HN indazol-1-yl)acetate (4f): In a 100 mL three necked round bottom flask, 2-methyl-4-nitroaniline (1f) (10 mmol) and citric acid (20 mmol) are dissolved in a mixture of ethanol (8 mL) and water (4 mL). The clear solution is cooled at 0-5°C and stirred well. To this added ice-cold solution of NaNO2 (2) (12 mmol) in 4 mL of water in 4 portions for 5-10 minutes. The formation of 1H-indazole is monitored by TLC. After the completion of the reaction as per TLC, ethyl chloroacetate (3) (12.0 mmol) is added to the reaction mixture and is stirred under reflux conditions for 60 min. The progress of the reaction is monitored by TLC. After completion of the reaction as per TLC, iron powder (30 mmol) and CaCl2 (10 mmol) are added to the reaction mixture and stirred at 60-65°C for 60 min. The progress of the reaction is monitored by TLC. After completion of the reaction as per TLC, reaction mixture is filtered to remove the iron residue and washed with EtOAc (2 × 10 mL). The organic layer is washed with water, dried over MgSO4 and concentrated. The obtained crude product (4f) is purified by silica gel using n-hexane and EtOAc (3:1 ratio). The same procedure is followed for the preparation of compounds 4g-j. The synthesized compounds (4f-j) gave satisfactory spectroscopic data in accordance with their proposed structures.
Results and Discussion
In the present study, our initial efforts are focused on identifying the catalyst, solvent(s) for the construction of 1H-indazole followed by N-alkylation in a single step operation. For this purpose, the reaction of 2-methyl anilines (1a) with NaNO2 (2) and followed by treatment with ethyl chloroacetate (3) in onepot was chosen as a model reaction to optimize the reaction conditions and the obtained results are presented in Table 1. The construction of 1H-indazole followed by N-alkylation involved in the present process which is carried out in the presence of various biodegradable α-hydroxy acidic catalysts such as malic acid, mandelic acid, tartaric acid and citric acid (1.0 equivalent) in water for about 150 min. Low yields of product 4a, 5%, 5%, 8% and 15%, are obtained respectively. To improve the yield, the same reaction is repeated by employing various solvents, like methanol, isopropanol, EtOH, EtOH:H2O (1:1 ratio), EtOH:H2O (2:1 ratio) and EtOH:H2O (3:1 ratio) and the results are presented in Table 1. After the examination of various α-hydroxy acid catalysts and solvents, it is found that citric acid (1.0 eq) and 1:1 ratio of EtOH:H2O medium is the best option to obtain good yield (75%) of the product (4a) and other α-hydroxy acid catalysts and solvents give low yields of product (4a).
| Entry |
Catalyst |
Solvent |
Product |
Time
(min) |
Yieldb
(%) |
| 1 |
Malic acid |
H2O |
4a |
120 |
5 |
| Methanol |
|
120 |
10 |
| Ethanol |
|
120 |
20 |
| Isopropanol |
|
120 |
15 |
| Ethanol:H2O (1:1 ratio) |
|
70 |
40 |
| Ethanol:H2O (2:1 ratio) |
|
70 |
35 |
| Ethanol:H2O (3:1 ratio) |
|
70 |
30 |
| 2 |
Mandelic acid |
H2O |
4a |
120 |
5 |
| Methanol |
|
120 |
10 |
| Ethanol |
|
120 |
25 |
| Isopropanol |
|
120 |
15 |
| Ethanol:H2O (1:1 ratio) |
|
70 |
45 |
| Ethanol:H2O (2:1 ratio) |
|
70 |
35 |
| Ethanol:H2O (3:1 ratio) |
|
70 |
30 |
| 3 |
Tartaric acid |
H2O |
4a |
120 |
10 |
| Methanol |
|
120 |
15 |
| Ethanol |
|
120 |
30 |
| Isopropanol |
|
120 |
20 |
| Ethanol : H2O (1:1 ratio) |
|
70 |
60 |
| Ethanol : H2O (2:1 ratio) |
|
70 |
55 |
| Ethanol : H2O (3:1 ratio) |
|
70 |
50 |
| 4 |
Citric acid |
H2O |
4a |
120 |
15 |
| Methanol |
|
120 |
20 |
| Ethanol |
|
120 |
40 |
| Isopropanol |
|
120 |
35 |
| Ethanol:H2O (1:1 ratio) |
|
70 |
75 |
| Ethanol:H2O (2:1 ratio) |
|
70 |
70 |
| Ethanol:H2O (3:1 ratio) |
|
70 |
65 |
aReaction conditions: 2-methyl aniline (1a) (10 mmol), NaNO2 (2) (12 mmol), citric acid (15 mmol), 2-ethyl chloroacetate (3) (10 mmol) in a mixture of Ethanol:H2O (1:1 ratio). bIsolated yield
Table 1: Screening of catalyst for the regioselective synthesis of ethyl 2-(1H-indazol-1-yl)acetate (4a) in one-pot.a
Effect of mode of addition of NaNO2
It is interesting to mention the effect of mode of NaNO2 addition. It is investigated and found that the portion wise addition and one time addition of NaNO2 had a significant effect on the isolated yield of the desired product (4a). For example, the isolated yield of the product (4a) is increased on addition of NaNO2 in portion wise (entries 2-4, Table 2) when compared to one-time addition of NaNO2 (entry 1, Table 2). The review concluded that addition of NaNO2 in 4 portions provided improved yield of product 4a (entry 4).
| Entry |
Mode of NaNO2
addition |
Time
(min) |
Product |
Yieldb
(%) |
| 1 |
Once |
20 |
4a |
75 |
| 2 |
2 portions |
10 |
4a |
80 |
| 3 |
3 portions |
10 |
4a |
87 |
| 4 |
4 portions |
10 |
4a |
96 |
aReaction conditions: 2-methyl aniline (1a) (10 mmol), NaNO2 (2) (12 mmol), (portion wise) citric acid (15 mmol), ethyl chloroacetate (3) (10 mmol) in a mixture of Ethanol:H2O (1:1 ratio). bIsolated yield.
Table 2: Screening of mode of addition of NaNO2 on the regioselective synthesis of ethyl 2-(1H-indazol-1-yl)acetate (4a) in one-pota.
Further, it is noteworthy to mention that the substrate (1) contains – NO2 group and is reduced to – NH2 in the presence of Fe, CaCl2 at 60-65°C for 30-40 min in the same pot. This functional group transformation is useful for the development of indazole based new chemical entities of potential pharmacological interest.
Scope of the method
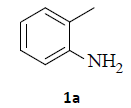
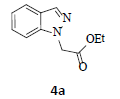
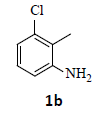
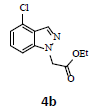
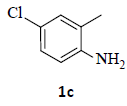
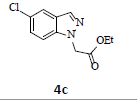
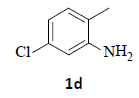
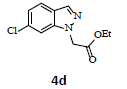
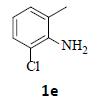
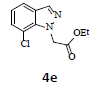
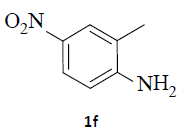
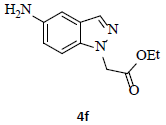
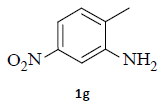
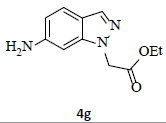
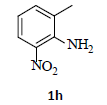
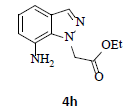
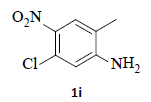
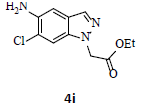
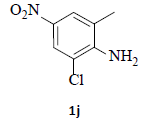
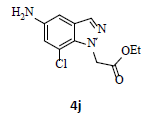
With the help of the above optimized reaction conditions, the scope of the present method is studied using different types of substituted 2-methyl anilines (1a-j) and the obtained results are presented in Table 3. The 2-methyl aniline (1a) provided excellent yields of desired product (4a) i.e. 96% (entry 1, Table 3). The substrates (1d) and (1c) bearing chloro group at 4-and 5- positions, afforded excellent yield of products 4c (94%) and 4d (94%) when compared to the same substituent at 2- and 6- positions of the substrates (1b) and (1e) (entries 2-5, Table 3). This may be due to the ortho effect. Substrate (1 g) with -NO2 group at 5-position, afforded good yields of product (4 g) when compared to the same substituent at 4- and 6- positions of the substrates (1f) and (1h) (entries 6-8, Table 3). The main reason is that the -NO2 group in (1f) and (1h) is suitably located for withdrawing of electrons strongly from NH2 group whereas this type of situation is not observed in the case of (1 g). Substrates (1i) and (1j) containing both nitro (– NO2) and chloro (– Cl) groups provided lower yields of products 4i (80%) and 4j (78%) compared to the mono substituted substrates (entries 9 and 10, Table 3). This may be attributed to the steric effects. From the above study, it is found that the substrates bearing weak electron-withdrawing substituents (i.e. chloro) (1b-e) provided excellent isolated yields of desired products in the range of 90- 94% (entries 2-5, Table 3) when compared to the substrates with strong electron-withdrawing substituents (i.e. nitro) (1f-h) (84- 92% yields) (entries 6-8, Table 3).
| Entry |
Substrate
(1) |
Product
(4) |
Time
(min) |
Yieldb
(%) |
| 1 |
 |
 |
70 |
96 |
| 2 |
 |
 |
80 |
90 |
| 3 |
 |
 |
75 |
94 |
| 4 |
 |
 |
75 |
94 |
| 5 |
 |
 |
90 |
92 |
| 6c |
 |
 |
120 |
86 |
| 7c |
 |
 |
100 |
92 |
| 8c |
 |
 |
120 |
84 |
| 9c |
 |
 |
130 |
80 |
| 10c |
 |
 |
130 |
78 |
aReaction conditions: 2-methyl anilines (1) (10 mmol), NaNO2 (2) (12 mmol), citric acid (20 mmol), 2-ethyl chloroacetate (3) (10 mmol) in a mixture of Ethanol : H2O (1:1 ratio). bIsolated yield cIf R''=NO2, the reduction process is carried out in the presence of Fe, CaCl2 at 60-65°C for 30-40 min in the same pot.
Table 3: One-pot, three-component regioselective synthesis of substituted 1H-indazole derivatives (4a-j)a.
Physical and spectral data of the synthesized compounds
1. Ethyl 2-(1H-indazol-1-yl)acetate (4a): Colourless oil, Yield=96%, 1H NMR (300 MHz): δ 8.25 (d, J=7.2 Hz, 1H, arom H), δ 8.0 (s, 1H, arom H), 7.80(d, J=7.2 Hz, 1H, arom H), 7.50-7.42 (m, 1H, arom H), 7.34-7.32 (m, 1H, arom H), 5.2 (s, 2H, N-CH2-) 4.22 (q, J=6.3Hz, 2H, -OCH2-), 1.24 (t, J=6.3Hz, 3H, CH3). MS (ESI): m/z [M+Na]+ 227.44.
2. Ethyl 2-(4-chloro-1H-indazol-1-yl)acetate (4b): Colourless oil, Yield=90%, 1H NMR (300 MHz): δ 8.07 (s, 1H, arom H), 7.65 (d, J=7.2Hz, 1H, arom H), 7.36 (d, J=7.2Hz, 1H, arom H), 7.10 (t, J=7.2Hz, 1H, arom H), 5.54 (s, 2H, -N-CH2-C=O), 4.23 (q, J=6.6Hz, 2H, -OCH2-), 1.26 (t, J=6.6Hz, 3H, CH3). MS (ESI): m/z [M+H+35Cl]+ 239.26; [M+H+37Cl]+ 241.28
3. Ethyl 2-(5-chloro-1H-indazol-1-yl)acetate (4c): Colourless oil, Yield=94%, 1H NMR (300 MHz): δ 8.05 (s, 1H, arom H), 7.72 (d, J=2.4Hz, 1H, arom H), 7.46 (d, J=6.6Hz, 1H, arom H), 5.14 (s, 2H, -N-CH2-C=O), 4.23 (q, J=6.9Hz, 2H, -OCH2- ), 1.27 (t, J=6.9Hz, 3H, CH3). MS (ESI): m/z [M+H+35Cl]+ 239.32; [M+H+37Cl]+ 241.36
4. Ethyl 2-(6-chloro-1H-indazol-1-yl)acetate (4d): Colourless oil, Yield=94%, 1H NMR (300 MHz): δ 8.04 (s, 1H, arom H), 7.68 (d, J=7.2Hz, 1H, arom H), 7.37 (s, 1H, arom H), 7.16 (d, J=7.2Hz, 1H, arom H), 5.12 (s, 1H, arom H), 4.24 (q, J=6.9Hz, 2H, -N-CH2-C=O), 1.28 (t, J=6.9Hz, 3H, CH3). MS (ESI): m/z [M+Na+] 261.24. [M+Na+35Cl]+ 261.24; [M+Na+37Cl]+263.24.
5. Ethyl 2-(7-chloro-1H-indazol-1-yl)acetate (4e): Colourless oil, Yield=92%, 1H NMR (300 MHz): δ 8.14 (s, 1H, arom H), 7.40-7.25 (m, 2H, arom H), 7.20 (d, J = 6.6Hz, 1H, arom H), 5.15 (s, 2H, -N-CH2-C=O), 4.22 (q, J=6.8Hz, 2H, -OCH2-), 1.25 (t, J=6.6Hz, 3H, CH3). MS (ESI): m/z 239.40. [M+H+35Cl]+239.40; [M+H+37Cl]+ 241.38
6. Ethyl 2-(5-amino-1H-indazol-1-yl)acetate (4f): Colourless oil, Yield=86%, 1H NMR (300 MHz): δ 7.85 (s, 1H, arom H), 7.14 (d, J=8.0Hz, 1H, arom H), 6.95 (s, 1H, arom H), 6.85 (d, J=8.0Hz, 1H, arom H), 5.1 (s, 2H, N-CH2-), 4.20 (q, J=6.0Hz, 2H, -OCH2-), 1.25 (t, J=6.0Hz, 3H, CH3). MS (ESI): m/z [M+Na+] 242.24.
7. Ethyl 2-(6-amino-1H-indazol-1-yl)acetate (4g): Colourless oil, Yield = 92%, 1H NMR (300 MHz): δ 7.75 (s, 1H, arom H), 7.50 (d, J=6.9Hz, 1H, arom H), 6.60 (d, J=6.9Hz, 1H, arom H), 6.50 (s, 1H, arom H), 5.0 (s, 2H, N-CH2-), 4.25 (q, J=6.3Hz, 2H, -OCH2-), 1.22 (t, J=6.3Hz, 3H, CH3). MS (ESI): m/z [M+H+] 220.22.
8. Ethyl 2-(7-amino-1H-indazol-1-yl)acetate (4h): Colourless oil, Yield = 84%, 1H NMR (300 MHz): δ 7.94 (s, 1H, arom H), 7.26 (d, J=6.3Hz, 1H, arom H), 6.96 (t, J=6.3Hz, 1H, arom H), 6.70 (d, J=6.3Hz, 1H, arom H), 5.4 (s, 2H, N-CH2-), 4.25 (q, J=6.6Hz, 2H,-OCH2-), 3.90 (br s, 2H, -NH2), 1.28 (t, J=6.6Hz, 3H, CH3). MS (ESI): m/z [M+H+] 220.40.
9. Ethyl 2-(5-amino-6-chloro-1H-indazol-1-yl)acetate (4i): Colourless oil, Yield = 80%, 1H NMR (300 MHz): δ 7.85 (s, 1H, arom H), 7.32 (s, 1H, arom H), 7.17 (s, 1H, arom H), 5.06 (s, 2H, -N-CH2-C=O), 4.22 (q, J=6.4Hz, 2H, -OCH2-), 1.27 (t, J=6.4Hz, 3H, CH3). MS (ESI): m/z [M+H+35Cl]+ 254.24; [M+H+37Cl]+ 256.30.
10. Ethyl 2-(5-amino-7-chloro-1H-indazol-1-yl)acetate (4j): Colourless oil, Yield = 78%, 1H NMR (300 MHz): δ 8.02 (s, 1H, arom H), 7.84 (s, 1H, arom H), 6.86 (d, J=2.4Hz, 1H, arom H), 5.42 (s, 2H, -N-CH2-C=O), 4.25 (q, J=6.9Hz, 2H, -OCH2-), 1.25 (t, J=6.9Hz, 3H, CH3). MS (ESI): m/z [M+Na+35Cl]+ 276.66; [M+Na+37Cl]+278.66.
Conclusion
A practical and green synthetic route has been developed for the regioselective synthesis of N-alkylated indazoles in onepot from readily available starting materials 2-methylanilines (1), NaNO2 (2) and ethyl chloroacetate (3) via diazotization, intra molecular cyclization and followed by N-alkylation in the presence of citric acid in a mixture of ethanol and water (1:1 ratio). Notably, if the substrate (1) contains – NO2 group and is reduced to – NH2 in the presence of Fe, CaCl2 in the same pot at 65°C. The superior features of the present method comprise of broad substrate scope, cleaner reaction profile, easy to perform, high yields of products and simple work-up procedure. Besides, the reaction is step-and atom-economic and is carried out in green catalytic medium. Thus, the method meets with the concepts of Wender’s “ideal synthesis”. Hence, we believe that this strategy would find applications in the synthesis of 1H-indazoles.
References
- Menor-Salván C, Martin-Yaseli MR (2012) Prebiotic chemistry in eutectic solutions at the water-ice matrix. Chemical Society Reviews 41: 5404-5415.
- Thangadurai A, Minu M, Wakode S, Agrawal S, Narasimhan B (2012) Indazole: a medicinally important heterocyclic moiety. Medicinal Chemistry Research 21: 1509-1523.
- Zhenxing L, Long W, Haocheng T, Shiyi Z , Tianren F, et al. (2014) Synthesis of 1H-indazoles from N-tosylhydrazones and nitroaromatic compounds. Chemical Communications 50: 5061-5063.
- Wyatt PG, Gill AL, Saxty G, Apaya R (2005) 1H-indazole-3-carboxamide compounds as MAPKAP kinase modulators. WO 2005014554.
- Arnold J, Artis DR, Hurt C, Ibrahim PN, Krupka H, et al. (2005) PPAR active compounds. WO 2005009958.
- May JA, Dantanarayana AP, Zinke PW, McLaughlin MA, Sharif NA (2006) 1-((S)-2-Aminopropyl)-1H-indazol-6-ol: A potent peripherally acting 5-HT2 receptor agonist with ocular hypotensive activity. Journal of Medicinal Chemistry 49: 318-328.
- Shimada I, Maeno K, Kazuta K, Kubota H, Kimizuka T, et al. (2008) Synthesis and structure-activity relationships of a series of substituted 2-(1H-furo[2,3-g]indazol-1-yl)ethylamine derivatives as 5-HT2C receptor agonists. Bioorganic Medicinal Chemistry 16: 1966-1982.
- Henke BR, Aquino CJ, Birkemo LS, Croom DK, Dougherty RW, et al. (1997) Optimization of 3-(1H-Indazol-3-ylmethyl)-1,5-benzodiazepines as potent, orally active CCK-A agonists. Journal of Medicinal Chemistry 40: 2706-2725.
- Schaus JM, Thompson DC, Bloomquist WE, Susemichel AD, Calligaro DO, et al. (1998) Synthesis and structure-activity relationships of potent and orally active 5-HT4 receptor antagonists: Indazole and Benzimidazolone derivatives. Journal of Medicinal Chemistry 41: 1943-1955.
- Vasudevan A, Souers AJ, Freeman JC, Verzal MK, Mathew JG, et al. (2005) Aminopiperidine indazoles as orally efficacious melanin concentrating hormone receptor-1 antagonists. Bioorganic Medicinal Chemistry Letters 15: 5293-5297.
- Lee CH, Brown BS, Perner RJ, Darbyshire J (2008) Indazole derivatives that inhibit trpv1 and uses thereof. WO 2008024945.
- Andronati S, Sava V, Makan S, Kolodeev G (1999) Synthesis of 3-aryl-1-[(4-phenyl-1-piperazinyl)butyl]indazole derivatives and their affinity to 5-HT1A serotonin and dopamine D1 receptors. Die Pharmazie 54: 99-101.
- Rodgers JD, Johnson BL, Wang H, Gienberg RA, Erickson VS, et al. (1996) Potent cyclic urea HIV protease inhibitors with benzofused heterocycles as P2/P2′ groups. Bioorganic & Medicinal Chemistry Letters 6: 2919-2924.
- Hartmann M, Sommer ME, Keppler BK, Kratz F, Einha¨user TJ (1995) New tumor-inhibiting ruthenium compounds: Investigations into their mode of action in human blood plasma and binding towards DNA. Journal of Inorganic Biochemistry 59: 214.
- Raffa D, Maggio B, Cascioferro S, Raimondi MV, Schillaci D, et al. (2009) Synthesis and antiproliferative activity of 3-amino-N-phenyl-1H-indazole-1-carboxamides. European Journal of Medicinal Chemistry 44: 165-178.
- Caron S, Vazquez EA (2006) Versatile and Efficient Synthesis of Substituted 1H-IndazolesSynthesis 1999: 588-592.
- Lukin K, Hsu MC, Fernando D, Leanna MR (2006) Journal of Organic Chemistry 71: 8166-8172.
- Sun JH, Teleha CA, Yan JS, Rodgers JD, Nugiel DA (1997) Efficient Synthesis of 5-(bromomethyl)- and 5-(aminomethyl)-1-THP-indazole. Journal of Organic Chemistry 62: 5627-5629.
- Chou LC, Huang LJ, Yang JS, Lee F, Teng CM, et al. (2007) Synthesis of furopyrazole analogs of 1-benzyl-3-(5-hydroxymethyl-2-furyl)indazole (YC-1) as novel anti-leukemia agents. Bioorganic &. Medicinal Chemistry 15: 1732-1740.
- Jennings A, Tennant M (2007) Selection of molecules based on shape and electrostatic similarity: proof of concept of electroforms. Journal of Chemical Information and Modeling 47: 1829-1838.
- Kingsbury WD, Gyurik RJ, Theodorides VJ, Parish RC, Gallagher G (1976) Synthesis of 1- and 2-substituted indazoles as anthelmintic agents. Journal of Medicinal Chemistry 19: 839-840.
- Palazzo G, Corsi G, Baiocchi L, Silvestrini B (1966) Synthesis and pharmacological properties of 1-substituted 3-dimethylaminoalkoxy-1H-indazoles. Journal of Medicinal Chemistry 9: 38-41.
- Shutske GM, Allen RC, Foersch MF, Setescak LL, Wilker JC (1983) Heterocyclic oxyacetic acid diuretics: indazole, benzisothiazole, and benzisothiazole 1,1-dioxide analogs of [[7-chloro-3-(2-fluorophenyl)-1,2-benzisoxazol-6-yl]oxy]acetic acid. Journal of Medicinal Chemistry 26: 1307-1311.
- Kovach EG, Barnes DE (1954) Preparation and properties of some new chelating agents. Journal of American Chemical Society 76: 1176-1178.
- Crestey F, Collot V, Stiebing S, Rault S (2006) A new and efficient synthesis of 2-azatryptophans. Tetrahedron 62: 7772-7775.
- Bartsch RA, cYang IW (1984) Phase transfer catalyzed synthesis of indazoles from o-alkylbenzenediazonium tetrafluoroborates. Journal of Heterocyclic Chemistry 21: 1063-1064.
- Magano J, Waldo M, Greene D, Nord E (2008) The synthesis of (S)-5-Fluoro-1-(2-fluorophenyl)-3-(piperidin-3-ylmethoxy)-1H-indazole, a Norepinephrine/Serotonin reuptake inhibitor for the treatment of Fibromyalgia. Organic Process Research & Development 12: 877-883.
- Kaïm LE, Grimaud L, Srinivas RP (2012) Four-component synthesis of Indazole through Ugi-Azide coupling. Synlett 2: 295-297.
- Ott GR, Anzalone AV (2011) Synthetic studies toward 3-(Acylamino)-1H-indazoles and development of a one-pot, Microwave-assisted, oxadiazole condensation/Boulton-Katritzky rearrangement. Synlett 20: 3018-3022.
- Lohou E, Collot V, Stiebing S, Rault S (2011) Direct access to 3-Aminoindazoles by Buchwald-Hartwig C-N coupling reaction. Synthesis 16: 2651-2663.
- Baraldi PG, Cacciari B, Spalluto G, Romagnoli R, Braccioli G, et al. (1997) A new synthetic approach to Indazole synthesis. Synthesis 10: 1140-1142.
- Ding QS, Zhang JL, Chen JX, Liu MC, Ding JC, et al. (2012) Tandem synthesis of 2,3-dihydroquinazolin-4(1H)-ones on grinding under solvent-free conditions. Journal of Heterocyclic Chemistry 49: 375-380.
- Spiteri C, Keeling S, Moses JE (2010) New Synthesis of 1-substituted-1H-indazoles via 1,3-dipolar cycloaddition of in situ generated nitrile imines and benzyne. Organic Letters 12: 3368-3371.
- Jin T, Yamamoto Y (2007) An efficient, facile, and general synthesis of 1H-Indazoles by 1,3-dipolar cycloaddition of arynes with diazomethane derivatives. Angewandte Chemie International Edition 46: 3323-3325.
- Yeu JP, Yeh JT, Chen TY, Uang BJ (2001) An expedient synthesis of 1-[3-(dimethylamino)propyl]-5-methyl-3-phenyl-1H-indazole (FS-32)-An antidepressant. Synthesis 12: 1775-1777.
- Liu Z, Shi F, Martinez PDG, Raminelli C, Larock RC (2008) Synthesis of Indazoles by the [3+2] cycloaddition of diazo compounds with arynes and subsequent acyl migration. Journal of Organic Chemistry 73: 219-226.
- Zhenqi Z, Tongsheng X, Xiaonai C, Yuzhu Q, Zheng Z (1993) A new and facile synthesis of 1H-indazoles. Journal of the Chemical Society, Perkin Transactions 1: 1279-1280.
- Vasconcelos AD, Oliveira PS, Ritter M, Freitag RA, Romano RL, et al. (2012). Antioxidant capacity and environmentally friendly synthesis of dihydropyrimidin-(2H)-ones promoted by naturally occurring organic acids. Journal of Biochemical and Molecular Toxicology 26: 155-161.
- Thopate SR, Kote SR, Rohokale SV, Thorat NM (2011) Citric acid catalysed Beckmann rearrangement, under solvent free conditions. Journal of Chemical Research 35: 124-125.
- Ghorbani-Choghamarani A, Taghipour T (2011) Green and One-Pot Three-Component Synthesis of 2,3-Dihydroquinazolin- 4(1H)-Ones Promoted by Citric Acid as Recoverable Catalyst in Water. Letters in Organic Chemistry 8: 470-476.
- Enugala R, Nuvvula S, Kotra V, Varala R, Adapa SR (2008) Green Approach for the Efficient Synthesis of Quinolines Promoted by Citric Acid. Heterocycles 75: 2523-2533.