Excessive angiogenesis (i.e. neovascularization) in
atherosclerotic lesions, sites of dissociation of the
inflammatory biomarker pentameric C-reactive protein
(pCRP) into monomeric CRP (mCRP), represents a focus of
plaque instability with haemorrhagic complications. We
previously demonstrated mCRP pro-angiogenic effects on
cultured aortic endothelial cells. However, mCRP effects in
combination with FGF-2, pro-angiogenic factor released by
activated macrophages infiltrating developing lesions, have
not yet been described. Here, we examined in vitro the
angiogenic capabilities of mCRP combined with FGF-2 by
performing endothelial cell proliferation, migration, and
differentiation including tube formation and spheroid
sprouting assays. The signaling pathways were also
investigated by Western blotting and all the cell-based
assays were used with or without pharmacological
inhibitors of mitogen-activated protein kinase (MAPK),
phosphatidylinositol-3 kinase (PI3K) and γ-secretase,
considered as key regulators of angiogenesis. We showed
that mCRP-induced endothelial cell proliferation and
migration required activation of PI3K pathway. MAPK
pathway was essential in mCRP-induced endothelial cell
proliferation and spheroid sprouting while γ-secretase
activity was indispensable for mCRP-induced tube formation
only. MAPK pathway was required in all FGF-2-stimulated
angiogenic assays whereas γ-secretase slightly inhibited
FGF-2 angiogenic effects. PI3K pathway was necessary for
FGF-2 angiogenic activities except for cell differentiation. In
most of the assays, the additive pro-angiogenic effects of
mCRP combined to FGF-2 were mainly attenuated by PI3K
and MAPK inhibitors. Altogether, mCRP and FGF-2 have
common angiogenic signaling pathways through PI3K and
MAPK. Thus, the therapeutic use of PI3K and MAPK
inhibitors may inhibit this increased vascularization whilst
reducing the haemorrhagic complications from unstable
plaques.
Keywords
Monomeric C-reactive protein; FGF-2; Angiogenesis; Signaling pathways; Inhibitors
Introduction
In Western and newly industrialized countries, atherosclerosis (complex chronic inflammatory disease) is the underlying cause of about 50% of all deaths related to stroke and cardio-vascular diseases [1,2]. Its incidence is continuing to rise as a result of the increase of potent risk factors for atherosclerosis including hypertension, obesity, type 2 diabetes, alcohol, and cigarette smoking [3-5]. In response to lipoprotein retention likely to mimic pathogen- and/or damage-associated molecular patterns, a low-grade inflammatory reaction is occurred in the atherosclerotic plaque through secretion of cytokines including growth factors such as basic fibroblast growth factor also known as FGF-2 and inflammatory biomarker such as C-reactive protein (CRP) produced by macrophage-derived foam cells and vascular cells [6-9]. It is noteworthy that both high level of expression of CRP and FGF-2 have been reported to be strongly correlated with atherosclerotic plaque instability [10,11]. In addition, approximately 75% of acute coronary events and 60% of symptomatic carotid artery disease are associated with disruption of atherosclerotic plaques [12].
Atherosclerotic plaque instability and vulnerability have been reported to occur through the activation of angiogenic process [13]. This angiogenic process target mainly endothelial cells, which play the major role in atherogenesis. Under the stimulation of the most potent angiogenic growth factor FGF-2, activated endothelial cells undergo metabolic modifications associated with the main steps of angiogenesis: i.e. production of matrix metalloproteinases (MMP) that degrade the basement membrane and extracellular matrix, stimulation of endothelial cell migration and proliferation; and differentiation resulting in sprout formation and ultimately, formation of new blood vessels [14,15]. These main FGF-2-induced angiogenic cell events occurred following to FGF-2-FGF receptor (FGFR) interactions that trigger a diverse array of cytoplasmic proteins of signaling pathways including mitogen-activated protein kinase (MAPK) such as extracellular-signal regulated kinase (ERK)1/2, the activation of phosphatidylinositol-3 kinase (PI3K) and involvement of γ-secretase, a transmembrane protein [16,17]. This intensified and uncontrolled angiogenic process contributes to the development of an unstable haemorrhagic rupture-prone environment [18].
C-reactive protein (CRP), inflammatory marker protein belonging to the family of pentraxins (five identical noncovalently linked subunits), is an acute-phase reactant and a strong independent predictor of cardio-vascular events [19-21]. CRP is physiologically constitutively synthesized in the liver at low rates whereas in pathological conditions associated with metabolic syndrome such as diabetes and atherosclerosis, CRP is predominantly produced by hepatic cells in response to inflammatory cytokines such as interleukin (IL)-6, IL-1β and tumour necrosis factor (TNF)- α released by fat cell adipocytes [22]. There are growing clinical trials and basic research evidence of the CRP as an active participant in the pathogenesis of atherosclerosis [23]. In addition, a dissociation of pentameric CRP (pCRP) into monomeric CRP (mCRP) occurs through its contact to the phosphatidylserine residue exposed on the surface of apoptotic cells and activated platelets, both cell entities located in the atherosclerotic plaque [24-27]. Several studies revealed that tissue-bound mCRP is the pro-bioactive component of the pCRP [28,29]. As a prognostic marker of the development and progression of vascular disease, mCRP plays a pivotal role by exerting direct modulatory effects on vascular cell functions including inflammatory, thrombotic, vasoactive, and angiogenic properties [30,31]. Although mCRP binds only to FcγRIII (CD16)-receptor bearing cells, its interactions with cholesterol-rich lipid rafts microdomains have been demonstrated to be the site of mCRP signaling in endothelial cells [32]. The pro-angiogenic activities of mCRP occur through activation of ERK/MAPK pathway, PI3K pathway and γ-secretase activity [31,33,34].
So far, the impact of the combination of mCRP with a potent pro-angiogenic factor such as FGF-2 has not yet been reported. In order to prevent the formation of unstable plaque with increasing hemorrhagic risks due to hyper-vascularization, there is a great interest to investigate the molecular mechanism underlying the combined pro-angiogenic effects of mCRP associated with FGF-2.
Materials and Methods
Reagents
Recombinant form of mCRP (0.5 mg/mL in 25 mM NaPBS, pH 7.4) was produced in the laboratory of Dr L.A. Potempa as previously described [35]. The mCRP solution contained an endotoxin concentration lower than 0.125 EU/mL and all cell culture medium was endotoxin-free. Recombinant human FGF-2 purified protein (18 kDa) was purchased from R&D systems (Abingdon, UK). All secondary antibodies conjugated to horseradish peroxidase were provided by DakoCytomation (Glostrup, Denmark). Rat tail collagen type I and growth factorreduced MatrigelTM were purchased from Becton Dickinson (Bredford, MA). Pharmacological inhibitors of MAPK pathway (PD98059), for PI3K pathway (LY294002) and for γ-secretase activity (DAPT) were provided by Santa Cruz biotechnology (Middlesex, UK). All other reagents and items were purchased from Sigma-Aldrich (Dorset, UK).
Culture of bovine aortic endothelial cells
Bovine aortic endothelial cells (BAEC) were isolated as previously described [36] and seeded in 75-cm2 flasks (NuncTM, Fisher Scientific, Loughborough, UK) pre-coated with 0.1% gelatine and cultured in Dulbecco’s Modified Eagle Medium (Lonza, Cambridge, UK) supplemented with 20% heatinactivated foetal bovine serum (FBS, Cambrex, Hertfordshire, UK), 2 mM glutamine and antibiotics (100 μg/mL streptomycin, 100 U/mL penicillin), defined as complete medium. The cells were incubated at 37°C in a saturated air humidity/5% CO2- incubator, passaged every 2-3 days, using enzymatic digestion with 0.05% trypsin/0.02% EDTA and split at a ratio of 1:2 or 1:3. At confluence, EC were identified by their typical cobblestone morphology. The cells were used throughout the study between passages 4 and 9.
Endothelial cell proliferation assay
BAEC (3 × 104 cells/well) were seeded in complete medium in 24-well plates (Nunc). After 4 h incubation, the medium was replaced with serum-poor medium (SPM; medium supplemented with 2.5% FBS) with or without 5 μg/mL mCRP and 25 ng/mL FGF-2, in the presence or absence of pretreatment (1 h) and co-treatment (72 h) of pharmacological inhibitors PD98059 (MAPK pathway inhibitor; 10 μM), LY294002 (PI3K pathway inhibitor; 10 μM), and DAPT (γ-secretase inhibitor; 10 μM). Each condition was performed in triplicate, with SPM or 0.05% dimethyl sulfoxide (DMSO)-treated cells used as negative controls. After 72 h incubation, the cells were detached in 250 μL of 0.05% trypsin / 0.02% EDTA and then each cell suspension was diluted in 10 mL of isotonic solution prior to counting using a Beckman-Coulter counter (London, UK).
Endothelial cell migration – wound recovery assay
BAEC (1.2 × 105 cells/well) were seeded in complete medium on Thermanox® coverslips (Nunc) in 24-well plates. After reaching pre-confluence following incubation for 24 h, each monolayer was wounded on two sides with a sterile razor blade, which gave the formation of two wound edges per coverslip with cell denuded areas. Dislodged cells were removed by washing with PBS without Ca2+/Mg2+. Then the coverslips were bathed in SPM with or without 5 μg/mL mCRP ± 25 ng/mL FGF-2 in the presence or absence of pre-treatment (1 h) and cotreatment (24 h) of pharmacological inhibitors PD98059 (MAPK pathway inhibitor; 10 μM), LY294002 (PI3K pathway inhibitor; 10 μM), and DAPT (γ-secretase inhibitor; 10 μM). Each condition was performed in duplicate and SPM or 0.05% DMSO-treated cells were used as negative controls. After 24 h incubation, the cells were washed with PBS, fixed with methanol and stained with 0.1% methylene blue for 2 min then washed abundantly with distilled water to reveal the wound recovery. Five pictures from the wound edge of each side were taken to assess the cell migration into the denuded area by counting the number of the migrated cells and by measuring the distance of the cell migration from the wound edge using Image J software (https:// rsb.info.nih.gov/nih.image/).
Endothelial tube formation assay in threedimensional Matrigel™ culture
BAEC (1.5 × 106 cells/mL) were mixed in equal volume with growth factor-reduced Matrigel™ (10 mg/mL) with or without 5 μg/mL mCRP ± 25 ng/mL FGF-2 in the presence or absence of pre-treatment (1 h) and co-treatment (24 h) of pharmacological inhibitors PD98059 (MAPK pathway inhibitor; 10 μM), LY294002 (PI3K pathway inhibitor; 10 μM), and DAPT (γ-secretase inhibitor; 10 μM). In 48-well plates (Nunc), each mixture was equally poured under a spot shape into two wells per experimental condition. After polymerisation of the gel for 1 h at 37°C, each spot of cells embedded in Matrigel™ was bathed in 500 μL of complete medium. Each condition was performed in duplicate and complete medium or 0.05% DMSO-treated cells were used as negative controls. After 24 h incubation, some cells migrated, aligned to form tubes with the formation of closed areas lined by the tubes. The cells were fixed with 4% paraformaldehyde for 15 min and the number of closed areas, used as a parameter for quantification of tubulogenesis [37], was counted in 5 random fields.
Three-dimensional sprout formation assay: spheroids embedded in collagen gel
BAEC culture spheroids were generated by mixing 125 μl of 3 × 105/mL of BAEC with 15 mL of 1.2% methylcellulose (4000 Cp viscosity). Then 150 μl of the 15 mL mixture were distributed in each well of 96-well Greiner® plates. After 2 days of incubation at 37°C and for each experimental condition, 48 well-formed spheroids were collected, centrifuged at 400 g for 3 min and resuspended in 250 μl of methylcellulose with or without either 5 μg/mL mCRP, 25 ng/mL FGF-2 or both in the presence or absence of pre-treatment (1 h) and co-treatment (24 h) of pharmacological inhibitors PD98059 (MAPK pathway inhibitor; 10 μM), LY294002 (PI3K pathway inhibitor; 10 μM), and DAPT (γ- secretase inhibitor; 10 μM). The collagen solution (250 μL of 2 mg/mL) which had been previously neutralised with 0.1 N NaOH was added to the spheroid mixture. Each collagen-spheroid mixture was equally distributed in a 24-well plate, in duplicate for each condition. After 2 days of incubation, sprouting occurred from the spheroid core and to measure the length of the sprouts, the spheroids were fixed with 4% paraformaldehyde for 15 min. The sprout length was estimated using the software Image J from 5 spheroids with a similar size core.
Collagen zymography
Matrix metalloproteinase (MMP) activities in the cell culture media which had been used for 3D sprout formation assay (as aforementioned described) were evaluated. The medium was collected and centrifuged (600 g for 15 min at 4°C). Protein concentration was determined using the Bradford protein assay (Bio-Rad, Munich, Germany), and the samples (100 μg of protein) were mixed with an equal volume of non-reducing sample buffer. The samples were incubated (10 min at room temperature) and subject to electrophoresis on 7.5% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) containing 2 mg/mL collagen I as a substrate. The gels were washed in renaturation buffer (2.5% Triton X-100) for 30 min at room temperature to remove SDS and to renature the MMPs in the gels. Then the gels were rinsed in activation buffer (50 mM Tris-HCl, 0.2 M NaCl, 5 mM CaCl2 and 0.02% Brij 35) for 30 min to activate the MMPs. Gels were incubated overnight at 37°C with fresh activation buffer then stained with 0.5% Coomassie blue R-250 for 2 h at room temperature. After destaining the gels, MMP collagenase activity was detected as a white band on a dark background and quantified by densitometry using Image J software.
Western blot analysis
For the study of the key signaling protein expression, BAEC (105 cells/well) were seeded in complete medium in a 24-well plate. After 48 h incubation, the medium was renewed with SPM for further 24 h incubation, following 1 h incubation of pretreatment with or without pharmacological inhibitors PD98059 (MAPK pathway inhibitor; 10 μM), LY294002 (PI3K pathway inhibitor; 10 μM), and DAPT (γ-secretase inhibitor; 10 μM) and then 5 μg/mL mCRP ± 25 ng/mL FGF-2 were added for 10 min stimulation at 37°C. Each condition was performed in duplicate and complete medium or 0.05% DMSO-treated cells were used as negative controls. After fast washing in cold PBS, cells were lysed with 120 μL/well of ice-cold radioimmunoprecipitation (RIPA) buffer (pH 7.5) as previously described until the transfer of the proteins onto nitrocellulose filters [31]. After protein estimation using Bradford protein assay (Biorad, Munchen, Germany), 100 μg of proteins were mixed with 2X Laemmli sample buffer then were separated by 10% SDS-PAGE and electroblotted (Hoefer, Bucks, UK) onto nitrocellulose filters (1 h). Nitrocellulose filters were blocked for 1 h at room temperature in TBS-Tween (pH 7.4) containing 1% BSA, and incubated with the following primary antibodies diluted in the blocking buffer, overnight at 4°C on a rotating shaker: rabbit monoclonal antibody to ERK1/2 (1:1000) and mouse monoclonal antibody to phospho-ERK1/2 (p-ERK1/2, 1:1000) were from Santa Cruz biotechnology. After washing (5 × 10 min in TBSTween at room temperature) filters were stained with either goat anti-rabbit or rabbit anti-mouse horse-radish peroxidase- conjugated secondary antibodies diluted in TBS-Tween containing 5% de-fatted milk (1:1000, 1 h, room temperature) with continuous mixing. After a further 5 washes in TBS-Tween, proteins were visualized by ECL (Geneflow Ltd, Staffordshire, UK) chemiluminescent detection and the image captured by Syngene imaging system was analysed using GeneSnap software (Syngene, Cambridge, UK).
Statistics
Results are expressed as mean ± standard deviation (SD). Experimental points were performed with a minimum of three independent experiments. A paired Student's t-test was used for comparison of two groups. A value of p<0.05 was considered significant.
Results
mCRP and FGF-2 increase EC proliferation, migration and tube formation
Tissue-bound monomeric C-reactive protein (mCRP) is an acute-phase reactant protein that is found co-located with high amount of the strongest pro-angiogenic factor FGF-2 in vulnerable atherosclerotic plaques [11,26].
Using endothelial cell-based angiogenesis assays including cell proliferation, migration, and cell differentiation, mCRP proangiogenic effects combined with FGF-2 were investigated. After 72 h of incubation, both 5 μg/mL mCRP (equivalent to a moderate acute-phase reactant) and 25 ng/mL FGF-2 increased the endothelial cell number by 1.3-fold (p=0.01) and 1.6-fold (p=0.0039), respectively, as compared to the untreated control cells (Figure 1A). A combination treatment of mCRP with FGF-2 showed additive effects (2.1-fold increase, p=0.0005) as compared to the control cells (Figure 1A).
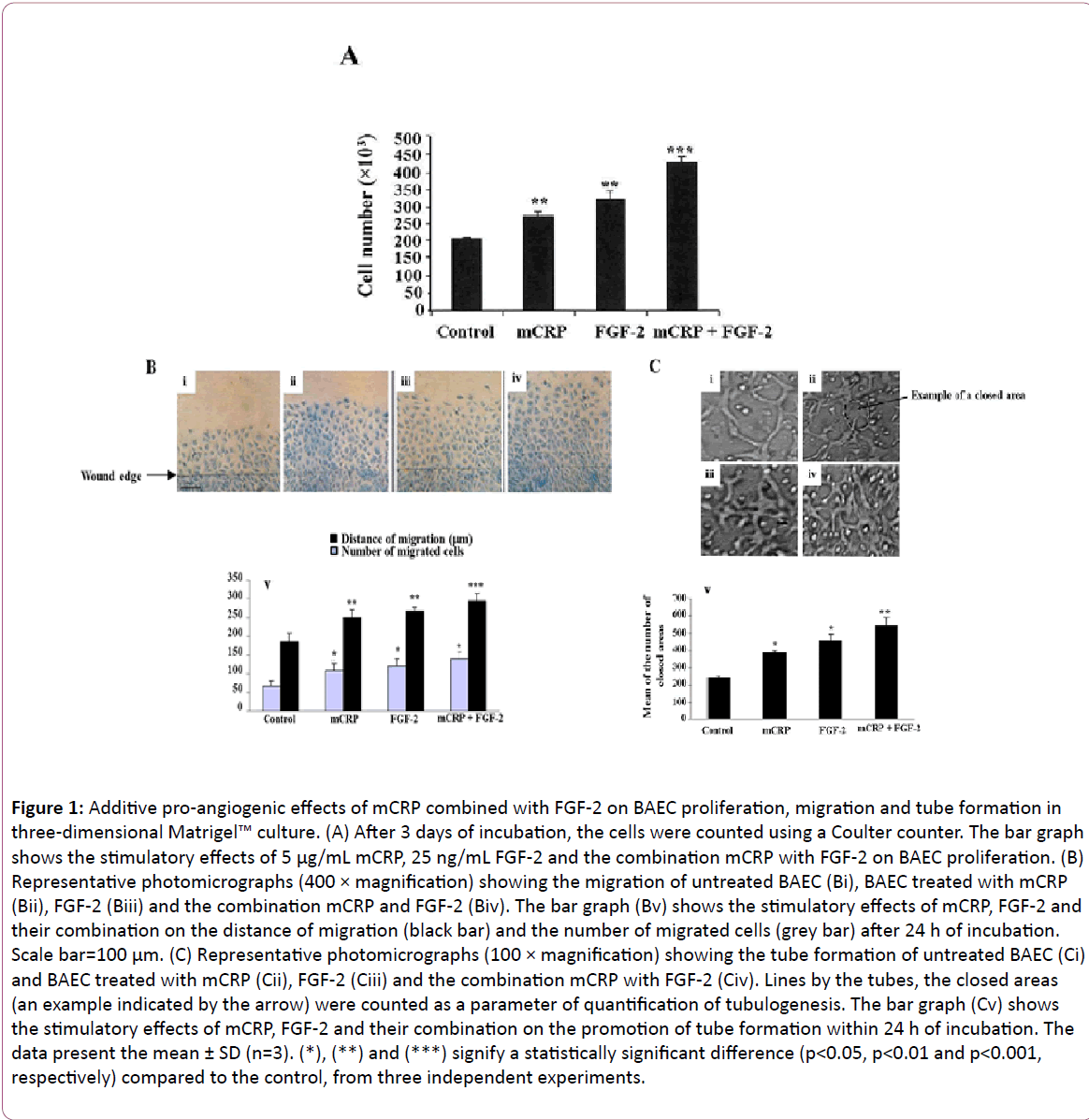
Figure 1: Additive pro-angiogenic effects of mCRP combined with FGF-2 on BAEC proliferation, migration and tube formation in three-dimensional Matrigel™ culture. (A) After 3 days of incubation, the cells were counted using a Coulter counter. The bar graph shows the stimulatory effects of 5 μg/mL mCRP, 25 ng/mL FGF-2 and the combination mCRP with FGF-2 on BAEC proliferation. (B) Representative photomicrographs (400 × magnification) showing the migration of untreated BAEC (Bi), BAEC treated with mCRP (Bii), FGF-2 (Biii) and the combination mCRP and FGF-2 (Biv). The bar graph (Bv) shows the stimulatory effects of mCRP, FGF-2 and their combination on the distance of migration (black bar) and the number of migrated cells (grey bar) after 24 h of incubation. Scale bar=100 μm. (C) Representative photomicrographs (100 × magnification) showing the tube formation of untreated BAEC (Ci) and BAEC treated with mCRP (Cii), FGF-2 (Ciii) and the combination mCRP with FGF-2 (Civ). Lines by the tubes, the closed areas (an example indicated by the arrow) were counted as a parameter of quantification of tubulogenesis. The bar graph (Cv) shows the stimulatory effects of mCRP, FGF-2 and their combination on the promotion of tube formation within 24 h of incubation. The data present the mean ± SD (n=3). (*), (**) and (***) signify a statistically significant difference (p< 0.05, p< 0.01 and p< 0.001, respectively) compared to the control, from three independent experiments.
Using wound healing assay, both mCRP (Figure 1Bii) and FGF-2 (Figure 1Biii) increased, after 24 h of incubation, the distance of migration by 1.3-fold (p=0.01) and 1.4-fold (p=0.01), respectively compared to the control cells (Figures 1Bi and 1Bv). Their combination (Figure 1Biv) increased the distance of migration by 1.5-fold (p=0.0009), compared to the control cells (Figure 1Bv). The number of migrated cells was increased by both mCRP (1.6-fold, p=0.037) and FGF-2 (1.8-fold, p=0.036), and even more by the combined factors (2.1-fold, p=0.023) as compared to the control cells (Figure 1Bv).
Endothelial cell differentiation was studied using a 3-D Matrigel™ culture. Within 24 h of incubation with respective treatments, activated cells migrated, aligned to form tubes organized in a capillary-like network (Figure 1C). Compared to the control (Figure 1Ci), mCRP (Figure 1Cii) and FGF-2 (Figure 1Ciii) increased the mean of closed areas lined by the tubes by 1.6-fold (p=0.02) and by 1.9-fold (p=0.03) respectively (Figure 1Cv). Moreover, the combined factors demonstrated an additive effect (2.2-fold, p=0.004, Figure 1Civ) with a significant difference when compared to each stimulus alone (Figure 1Cv).
mCRP and FGF-2 increase EC spheroid outgrowth associated with MMP-2/-9 activation
In order to further investigate the effects of mCRP and FGF-2 on the induction of the blood vessel morphogenesis, a sprouting angiogenesis assay was utilized to detect 3D-outgrowth of BAEC spheroids embedded in a collagen gel (Figure 2A).
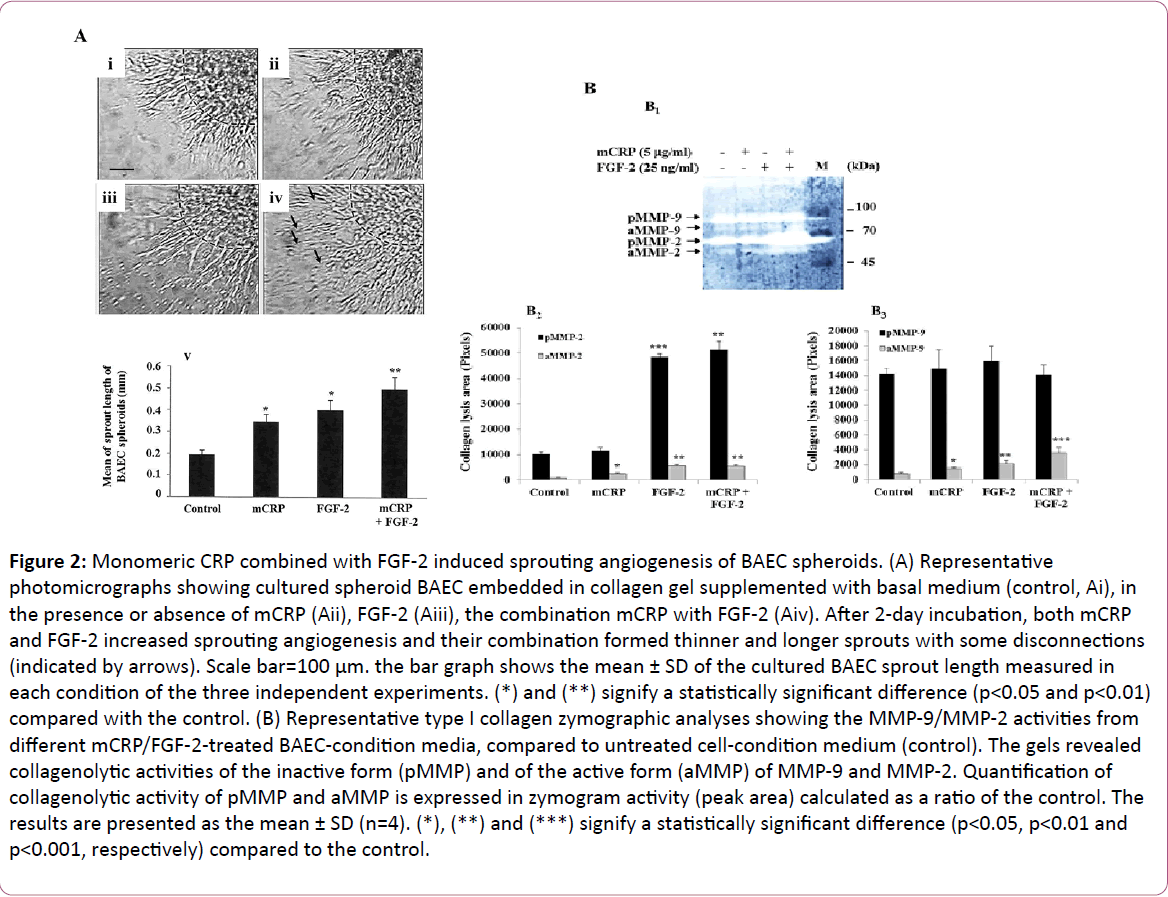
Figure 2: Monomeric CRP combined with FGF-2 induced sprouting angiogenesis of BAEC spheroids. (A) Representative photomicrographs showing cultured spheroid BAEC embedded in collagen gel supplemented with basal medium (control, Ai), in the presence or absence of mCRP (Aii), FGF-2 (Aiii), the combination mCRP with FGF-2 (Aiv). After 2-day incubation, both mCRP and FGF-2 increased sprouting angiogenesis and their combination formed thinner and longer sprouts with some disconnections (indicated by arrows). Scale bar=100 μm. the bar graph shows the mean ± SD of the cultured BAEC sprout length measured in each condition of the three independent experiments. (*) and (**) signify a statistically significant difference (p< 0.05 and p< 0.01) compared with the control. (B) Representative type I collagen zymographic analyses showing the MMP-9/MMP-2 activities from different mCRP/FGF-2-treated BAEC-condition media, compared to untreated cell-condition medium (control). The gels revealed collagenolytic activities of the inactive form (pMMP) and of the active form (aMMP) of MMP-9 and MMP-2. Quantification of collagenolytic activity of pMMP and aMMP is expressed in zymogram activity (peak area) calculated as a ratio of the control. The results are presented as the mean ± SD (n=4). (*), (**) and (***) signify a statistically significant difference (p< 0.05, p< 0.01 and p< 0.001, respectively) compared to the control.
Under the effect of each stimulatory agent, more cells migrated and aligned to form longer sprouts from the core of the spheroid. After 48 h of incubation and compared to the control, sprout length increased to the same extent by both mCRP (1.8-fold, p=0.03, Figure 2Aii) and FGF-2 (2.0-fold, p=0.04, Figure 2Aiii). However, a distinction was observed in the vascular structure in mCRP-treated cells showing fewer organisations and more filopodia with fewer interactions between the cells. Additive effects were observed when mCRP and FGF-2 were added together inducing an additional significant increase in the sprout length (2.5-fold, p=0.001, Figure 2Aiv) and less organisation as compared to the untreated control cells.
The outgrowth of the BAEC sprouts embedded in type I collagen occurs through the degradation of the collagen fibres carried out by the matrix metalloproteinases (MMPs). To assess and characterize the MMP activity involved in mCRP or FGF-2- induced EC spheroid outgrowth, collagen substrate zymography was performed. Collagen lysis areas corresponded to the molecular weight of the well-known gelatinases pro-MMP-9 and pro-MMP-2, which have been lately defined as type I collagenases [38,39]. Compared with MMP-2 activity assessed in control untreated BAEC-condition medium, both mCRP (2.63- fold, p=0.016, Figure 2B2) and FGF-2 (6.0-fold, p=0.001, Figure 2B2) increased the activity of MMP-2 (Figure 2B). No enhancement of MMP-2 activity from the combination mCRP with FGF-2-treated BAEC-condition medium was observed as compared with MMP-2 activity detected from FGF-2-treated BAEC-condition medium (Figure 2B). In regards to MMP-9 activity, both mCRP (1.88-fold, p=0.01, Figure 2B3) and FGF-2 (2.76-fold, p=0.006, Figure 2B3) increased MMP-9 collagenolytic activity, compared with MMP-9 assessed in control untreated BAEC-condition medium. The combination mCRP with FGF-2 (about 2.0-fold, p<0.01, Figure 2B3) enhanced the activity of MMP-9, compared with MMP-9 assessed in mCRP- or FGF-2- treated BAEC-condition medium (Figure 2B).
mCRP and FGF-2 increase ERK1/2 phosphorylation mainly through MAPK and PI3K pathways
We sought the existence of common converging angiogenic signaling pathways induced by both mCRP and FGF-2. To simplify this investigation of signaling pathways involved, we assessed the phosphorylation of extracellular signal-regulated kinase (ERK)1/2, the activated form of the most angiogenic signaling pathways downstream target, after endothelial cell treatment with mCRP and FGF-2, for 10 min incubation (optimal incubation time determined from pilot studies). Both mCRP and FGF-2 increased p-ERK1/2 expression by about 2.0-fold (p<0.05; Figure 3A), compared with the level of p-ERK1/2 detected in control untreated cells. The combined treatment with mCRP and FGF-2 was found to have an additive effect on p-ERK1/2 expression level showing a 4.0-fold (p=0.004 for p-ERK1, Figure 3A) and 5.0- fold (p=0.007 for p-ERK2, Figure 3A), compared to untreated control cells.
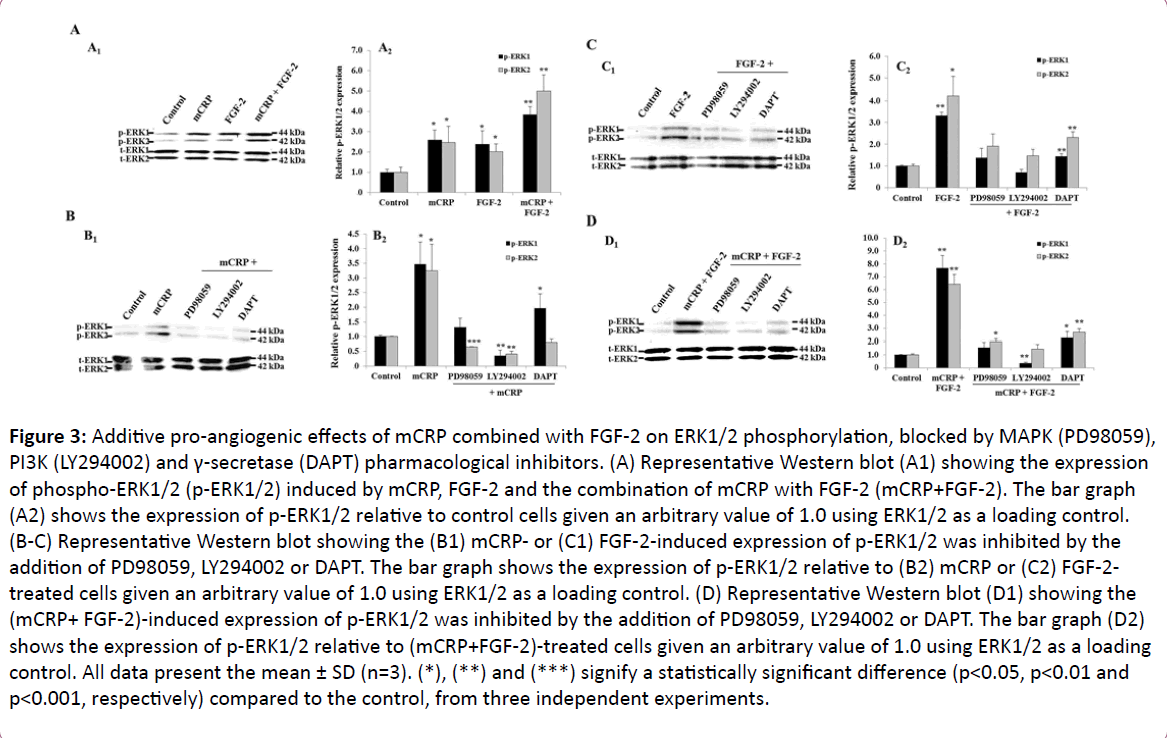
Figure 3: Additive pro-angiogenic effects of mCRP combined with FGF-2 on ERK1/2 phosphorylation, blocked by MAPK (PD98059), PI3K (LY294002) and γ-secretase (DAPT) pharmacological inhibitors. (A) Representative Western blot (A1) showing the expression of phospho-ERK1/2 (p-ERK1/2) induced by mCRP, FGF-2 and the combination of mCRP with FGF-2 (mCRP+FGF-2). The bar graph (A2) shows the expression of p-ERK1/2 relative to control cells given an arbitrary value of 1.0 using ERK1/2 as a loading control. (B-C) Representative Western blot showing the (B1) mCRP- or (C1) FGF-2-induced expression of p-ERK1/2 was inhibited by the addition of PD98059, LY294002 or DAPT. The bar graph shows the expression of p-ERK1/2 relative to (B2) mCRP or (C2) FGF-2- treated cells given an arbitrary value of 1.0 using ERK1/2 as a loading control. (D) Representative Western blot (D1) showing the (mCRP+ FGF-2)-induced expression of p-ERK1/2 was inhibited by the addition of PD98059, LY294002 or DAPT. The bar graph (D2) shows the expression of p-ERK1/2 relative to (mCRP+FGF-2)-treated cells given an arbitrary value of 1.0 using ERK1/2 as a loading control. All data present the mean ± SD (n=3). (*), (**) and (***) signify a statistically significant difference (p< 0.05, p< 0.01 and p< 0.001, respectively) compared to the control, from three independent experiments.
To demonstrate the involvement of angiogenic signaling pathways mediated by either mCRP or FGF-2, the EC were treated with pharmacological inhibitors blocking MAPK, PI3K pathways and γ-secretase named as PD98059, LY294002 and DAPT, respectively. From pilot studies, all pharmacological inhibitors and DMSO used did not change the low basal level of p-ERK1/2 detected in untreated cells, indicative of non-cytotoxic effect. An attenuation of mCRP (Figure 3B) or FGF-2 (Figure 3C)- induced ERK1/2 phosphorylation was observed by the cell pretreatment with the three pharmacological inhibitors (Figures 3B and 3C).
To highlight common pathways of this additive stimulatory effects triggered by mCRP combined with FGF-2, the EC were treated with the aforementioned pharmacological inhibitors. Each pharmacological inhibitor inhibited ERK1/2 phosphorylation induced by the combined factors mCRP and FGF-2; however, strong inhibitions were observed by the cell pre-treatment with either LY294002, the PI3K inhibitor or PD98059, the MAPK inhibitor (Figure 3D).
LY294002 and PD98059 attenuate mCRP and FGF-2 pro-angiogenic activities in vitro
Using the pharmacological inhibitors of PI3K and MAPK pathways, and of γ-secretase activity, we demonstrated that the cell pre-treatment with PD98059 (MAPK inhibitor) fully prevented FGF-2-stimulated BAEC proliferation (Figure 4A), migration (Figure 4B), tube formation (Figure 4C) and spheroid sprouting (Figure 4D), compared to respective control untreated cells. The cell pre-treatment with LY294002 (PI3K inhibitor) fully prevented FGF-2-induced cell proliferation and migration while LY294002 decreased FGF-2-induced cell differentiation such as tube formation by 51.5% (p=0.0017, Figure 4C) and spheroid sprouting by 37.2% (p=0.047, Figure 4D), as compared to FGF-2 alone. The blockade of γ-secretase using DAPT partially inhibited FGF-2 mitogenic effect (-36.86%, p=0.007, Figure 4A), while no significant inhibitory effect of FGF-2-induced cell migration, tube formation, and spheroid sprouting was observed, as compared to FGF-2 alone. In regards to the signaling pathways involved in mCRP pro-angiogenic effects, the blockade of PI3K pathway using LY294002 fully prevented mCRP-stimulated BAEC proliferation (Figure 4E) and migration (Figure 4F), while LY294002 inhibited mCRP-induced tube formation (-54.7%, p=0.035, Figure 4G) and spheroid sprouting (-35.7%, p=0.04, Figure 4G), as compared to mCRP alone (Figure 4). The blockade of MAPK pathway using PD98059 totally inhibited mCRP-induced BAEC proliferation (Figure 4E) and the number of migrated cells (Figure 4F) as compared to mCRP alone. PD98059 pre-treatment also decreased mCRP-induced tube formation (-46.3%, p=0.03, Figure 4G) and spheroid sprouting (-50.9%, p=0.001, Figure 4H) as compared to mCRP alone. The blockade of γ-secretase activity totally prevented mCRP-induced tube formation (Figure 4G) while a partial inhibition of mCRP-induced spheroid sprouting was observed (-40.3%, p=0.004, Figure 4H) as compared to mCRP alone.
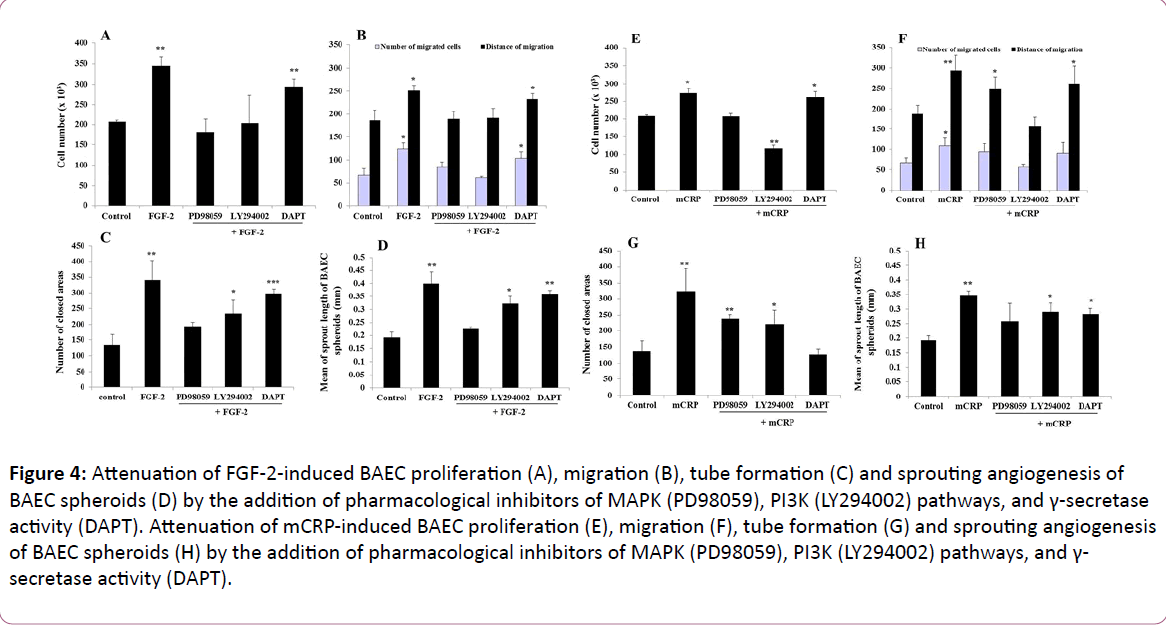
Figure 4: Attenuation of FGF-2-induced BAEC proliferation (A), migration (B), tube formation (C) and sprouting angiogenesis of BAEC spheroids (D) by the addition of pharmacological inhibitors of MAPK (PD98059), PI3K (LY294002) pathways, and γ-secretase activity (DAPT). Attenuation of mCRP-induced BAEC proliferation (E), migration (F), tube formation (G) and sprouting angiogenesis of BAEC spheroids (H) by the addition of pharmacological inhibitors of MAPK (PD98059), PI3K (LY294002) pathways, and γ- secretase activity (DAPT).
In order to highlight the signaling pathways required for the additive pro-angiogenic effects of the combination mCRP with FGF-2, we compared the impact of the different aforementioned pharmacological inhibitors on the angiogenic effects of the combined factors mCRP and FGF-2. The increased BAEC proliferation induced by the combination factors mCRP and FGF-2 was mainly attenuated by the blockade of both MAPK and PI3K pathways (Figure 5A). In regards to cell migration, the blockade of PI3K pathway totally inhibited the combined factors (mCRP and FGF-2)-induced cell migration, while this combined stimulatory effect was not reduced after the blockade of MAPK pathway and γ-secretase activity (Figure 5B). In regards to endothelial cell differentiation, both MAPK and γ-secretase inhibitors mainly prevented the combined factors (mCRP and FGF-2)-induced tube formation, compared to the tube formation induced by the combined factors mCRP and FGF-2 without pharmacological inhibitors (Figure 5C). In regards to sprouting angiogenesis, the blockade of MAPK, PI3K pathways and γ- secretase activity decreased the combined factors (mCRP and FGF-2)-induced BAEC sprouting by 78% (p=0.02), 49% (p=0.02) and 33% (p=0.03), compared to BAEC sprouting induced by the combined factors mCRP and FGF-2 without pharmacological inhibitors, respectively (Figure 5D).
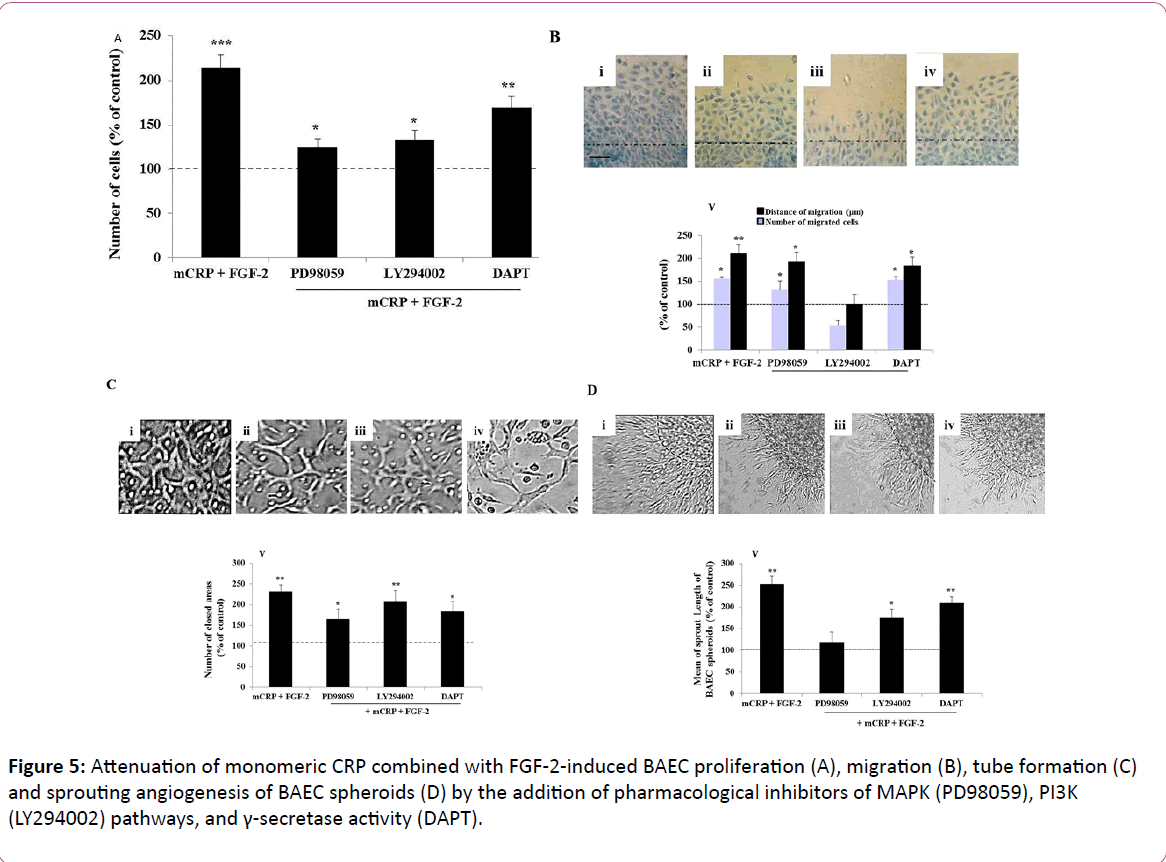
Figure 5: Attenuation of monomeric CRP combined with FGF-2-induced BAEC proliferation (A), migration (B), tube formation (C) and sprouting angiogenesis of BAEC spheroids (D) by the addition of pharmacological inhibitors of MAPK (PD98059), PI3K (LY294002) pathways, and γ-secretase activity (DAPT).
Discussion
Atherosclerosis remains a high-cost underlying pathogenic component of cardiovascular disease, and its complications still represent the primary cause of death in most industrialized countries [3]. Various inflammatory mediators and cytokines including mCRP and FGF-2 have been detected in unstable atherosclerotic lesion formation and were associated with regions of a vascularization leading to increased risk of microvessel haemorrhage [11,18,40]. The main interest of this study was to get a better understanding of the in vitro pro-angiogenic effects of the combined factors mCRP with FGF-2 and of the associated signaling pathways targeting ERK1/2, the main key regulator of angiogenic cellular and molecular processes. Using various pharmacological inhibitors blocking MAPK, PI3K pathways and γ-secretase activity, we have demonstrated that MAPK and PI3K pharmacological inhibitors mostly fully decreased the angiogenic processes induced by the combined factors mCRP with FGF-2; suggesting that mCRP and FGF-2 have common signaling pathways engaging mainly PI3K and MAPK. Thus, the therapeutic use of PI3K and MAPK inhibitors might have the potential to inhibit this increased vascularization whilst reducing the haemorrhagic complications from unstable plaques.
Here, we showed that both mCRP and FGF-2 increased aortic endothelial cell proliferation, migration, tube formation and sprouting remodelling abilities. These results confirmed previous studies using similar or other origins of endothelial cells [28,31,41]. The angiogenic endothelial cell response was additively enhanced after cell exposure to mCRP combined with FGF-2 as compared with single mCRP or FGF-2 stimulation. This additive angiogenic effect mediated by the combination mCRP and FGF-2 was also confirmed by the over-phosphorylation of ERK1/2, of which the level reached was higher than the ERK1/2 phosphorylation level obtained under single stimulation. In this present study, the investigation of the signaling pathways activated by either mCRP, FGF-2 or their combination allowed us to highlight any potential common molecular mechanisms that could explain the combination (mCRP with FGF-2)-induced additive angiogenic cellular processes.
In regards to cell proliferation, the cell pre-treatment with either MAPK inhibitor (PD98059) or PI3K inhibitor (LY294002) fully prevented mCRP and FGF-2 mitogenic activities as well as their combined activities, suggesting the involvement of two main distinct signaling pathways through GRB2, a FGFR docking protein that recruits the guanine nucleotide exchange factor SOS and the adaptor protein GAB1, which are involved in ERK1/2 MAPK pathway and PI3K pathway activation, respectively [42]. Activated ERK1/2 translocates to the nucleus and transactivates transcription factors such as Ets while Akt phosphorylates and inactivates transcription factor such as forkhead box O (FOXO1), changing gene expression to promote growth or mitosis [43,44]. In contrast to FGF-2 that mainly acts through tyrosine kinase receptor FGFR, mCRP activates endothelial cells through cholesterol-rich lipid raft microdomains, sites of phosphorylation of signaling proteins including ERK1/2 and PI3K/Akt [45,46]. In addition, a direct association of lipid rafts with γ-secretase activity has also been revealed [47]. In this present study, γ- secretase inhibitor slightly decreased FGF-2 and mCRP mitogenic effects. Various studies have reported intertwined molecular interactions between ERK1/2, PI3K and γ-secretase activity. ERK1/2 has been shown to be an endogenous negative regulator of the γ-secretase activity by phosphorylating nicastrin, an essential component of γ-secretase; while presenilin, another key component of γ-secretase activity has been shown to be able to activate PI3K and ERK1/2 [48,49]. Using ductus ateriosus smooth muscle cells, Wu and colleagues demonstrated that DAPT inhibits cell proliferation by not only inhibiting ERK1/2 and Akt phosphorylation but also by promoting cell cycle arrest in the G0/G1–phase [50]. Collectively, these effects of single stimulation of mCRP, FGF-2 and their combination mCRP with FGF-2 on EC proliferation confirm the theory that growth factors display additive and even synergistic characteristics during early stages of angiogenesis, with the combination of at least two growth factors displaying the highest proliferation rate [51].
Using wound healing assay, we showed an additive increase in the number of migrated BAEC and in the distance of migrated cells from the wound edge in the presence of FGF-2 combined with mCRP, compared to single stimulation. Both FGF-2 and CRP have previously been demonstrated as potent promoters of cell motility of vascular cells including EC and smooth muscle cells [29,52-54]. EC migration is an essential component of angiogenesis that requires a tight mechanically integrated molecular process, which is associated with the activation of intracellular pathways such as PI3K and phosphorylation of focal adhesion kinase (FAK) that converge on cytoskeleton remodelling. In this present study, by blocking either MAPK or PI3K pathway, a significant reduction of FGF-2-induced BAEC migration was observed. However, the blockade of γ-secretase activity did not affect FGF-2-induced cell migration. The importance of MAPK and PI3K signaling pathways in FGF-2- induced endothelial cell migration was also confirmed by previous studies [55,56]. Here, PI3K blockade results in a full inhibition of BAEC migration induced by mCRP, while mCRPinduced distance of cell migration was remained unaffected after either MAPK or γ-secretase blockade. The additive effects of mCRP combined with FGF-2 were completely blocked by PI3K inhibitor, suggesting PI3K pathway as a shared common signaling pathway involved in the induction of endothelial cell migration [14,57].
Following migration, cell differentiation occurs to contribute to vessel maturation. In this present study, to mimic this last angiogenic step, BAEC were embedded either in Matrigel™ displaying tube formation or in type I collagen gel forming outgrowth from spheroids with the studied pro-angiogenic factors mCRP and FGF-2. Both mCRP and FGF-2 enhanced cell differentiation based on tube formation and spheroid sprouting. The combination of mCRP with FGF-2 additively increased both endothelial cell differentiation models as compared with single stimulation and was associated with an enhancement of MMP-9 collagenolytic activity. MMP-9 represents one of the key MMPs involved in the degradation of the connective tissue microenvironment for vascular remodelling and reported to be produced via MAPK and PI3K/Akt signaling pathways and requiring γ-secretase activity [58,59]. In addition, in this present study, we observed an increase of MMP-2 and MMP-9 activities after cell stimulation with either FGF-2 or mCRP. Both FGF-2 and (m)CRP have been reported to stimulate the MMP-2/-9 production and their proteolytic activities using endothelial cells, smooth muscle cells and macrophages [60-62]. In addition, high proteolytic activity of MMP-9 has been demonstrated to be associated with plaque rupture while increased activity level of MMP-2 was associated with stable lesions, which reported that the MMPs activities differ among carotid plaque phenotype [62,63], supporting our findings showing that mCRP enhanced MMP-9 collagenolytic activity and reinforcing the mCRP causative role in unstable plaque [26]. FGF-2-induced tube formation was fully inhibited after the blockade of MAPK pathway while it was partially inhibited after the blockade of PI3K pathway. In alignment with our findings, both ERK1/2 MAPK and PI3K signaling pathways have been demonstrated to be required for the induction of cell differentiation [64]. In addition, in this present study, both MAPK and PI3K signaling pathways were shown to be partially involved in mCRP-induced tube formation. However, γ-secretase activity did not affect FGF-2-induced tube formation whereas, as previously shown, γ- secretase is essential for mCRP-induced tube formation [31]. Recently, mCRP-induced tube formation was demonstrated to activate F3 transcription factor and tissue factor signaling in microvascular endothelial cells [29]. As a consequence of additive combined effects, all pharmacological inhibitors of ERK1/2, PI3K pathways, and of γ-secretase activity partially inhibited the endothelial tube formation stimulated by mCRP associated with FGF-2. In regards to spheroid sprout structures, mCRP displayed disorganised sprout morphology in comparison to FGF-2. Using transmission electron microscopy, we previously showed that mCRP stimulated the formation of disorganised, nascent vasculatures containing high numbers of EC fenestrations, which might be associated with high vascular permeability, aligning with the risk of haemorrhage aforementioned [26,31]. Using similar spheroid assay, FGF-2 has produced longer aligned, organised and thicker sprouts than mCRP. Sustained and extensive FGF-2-induced neovascularization has been reported to cause haemorrhage [65]. The combination of mCRP and FGF-2 displayed longer, thicker, and sparser sprouts when compared to the control cells and to mCRP and FGF-2 alone. The morphogenesis effects of mCRP, FGF-2 or mCRP combined with FGF-2 was mainly attenuated after blocking MAPK and PI3K signaling pathways while these effects were partially inhibited after the blockade of γ-secretase activity. Additional studies are needed to determine whether our observations in regards with the potential risk of haemorrhage due to aggressive neovascularization induced by mCRP combined with FGF-2 through the use of mCRP combined with FGF-2-loaded matrigel plugs, for instance [65]. It would be also of a great interest to check whether the inhibition of common signaling pathway of mCRP and FGF-2 could retard the progression of instable plaques in vivo using animal models of atherosclerosis.
In conclusion, our results indicate that mCRP combined with FGF-2, both cytokines predominantly found in atherosclerotic plaque, vastly increase aortic endothelial cell proliferation, migration, differentiation including tube formation and spheroid sprouting associated with MMP-9 production, main cause of plaque rupture. The morphological appearance of BAEC spheroid sprouts were not only thick structures, but less organised with fewer interactions between the cells, mimicking the high risk of leak. The cell pre-treatment with pharmacological inhibitors blocking MAPK (PD98059), PI3K pathways (LY294002), and γ-secretase activity (DAPT) demonstrated that angiogenic cell response with associated signaling pathways mainly engaged MAPK and PI3K. Thus, we suggest that the combination of mCRP with FGF-2, main biomarkers of the unstable atherosclerotic plaque, leading to enhanced angiogenesis which is linked with increased risk of haemorrhage, might be prevented by blocking MAPK and PI3K signaling pathways.
Acknowledgment
We are grateful to the Libyan Ministry of Higher Education for providing a PhD scholarship to Emhamed Boras enabling him to undertake this study.
References
- Tabas I, García-Cardeña G, Owens GK (2015) Recent insights into the cellular biology of atherosclerosis. J Cell Biol 209: 13-22.
- Liu XY, Yan F, Niu LL, Chen QN, Zheng HR, et al. (2016) Strong correlation between early stage atherosclerosis and electromechanical coupling of aorta. Nanoscale 8: 6975-6980.
- Verma AK, Kumar S, Kumar N, Verma RK, Singh M (2012) Study of coronary artery atherosclerosis in sudden deaths and its medico-legal relevance. J Indian Acad Forensic Med 34: 132-134.
- Martín-Timón I, Sevillano-Collantes C, Segura-Galindo A, Del Cañizo-Gómez FJ (2014) Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J Diabetes 5: 444-470.
- World Health Organization (2014) Global status report on non-communicable diseases.
- Falcone DJ, McCaffrey TA, Haimovitz-Friedman A, Vergilio JA, Nicholson AC (1993) Macrophage and foam cell release of matrix-bound growth factors. Role of plasminogen activation. J BiolChem 268: 11951-11958.
- Chen CH, Jiang W, Via DP, Luo S, Li TR, et al. (2000) Oxidized low-density lipoproteins inhibit endothelial cell proliferation by suppressing basic fibroblast growth factor expression. Circulation 101: 171-177.
- Grad E, Pachino RM, Danenberg HD (2011) Endothelial C-reactive protein increases platelet adhesion under flow conditions. Am J Physiol Heart CircPhysiol 301: H730-736.
- Kaplan M, Hamoud S, Tendler Y, Meilin E, Lazarovitch A, et al. (2014) A significant correlation between C-reactive protein levels in blood monocytes derived macrophages versus content in carotid atherosclerotic lesions. J Inflamm (Lond.) 11: 7.
- Schwartz SM, Galis ZS, Rosenfeld ME, Falk E (2007) Plaque rupture in humans and mice. ArteriosclerThrombVascBiol 27: 705-713.
- Sigala F, Savvari P, Liontos M, Sigalas P, Pateras IS, et al. (2010) Increased expression of bFGF is associated with carotid atherosclerotic plaques instability engaging the NF-?B pathway. J Cell MolBiol 14: 2273-2280.
- Fishbein MC (2010) The vulnerable and unstable atherosclerotic plaque. CardiovascPathol 19: 6-11.
- Taruya A, Tanaka A, Nishiguchi T, Matsuo Y, Ozaki Y, et al. (2015) Vasa vasorum restructuring in human atherosclerotic plaque vulnerability: A clinical optical coherence tomography study. J Am CollCardiol 65: 2469-2477.
- Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, et al. (1998) Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An autocrine mechanism contributing to angiogenesis. J Cell Biol 141: 1659-16736.
- Doyle B, Caplice N (2007) Plaque neovascularization and antiangiogenic therapy for atherosclerosis. J Am CollCardiol 49: 2073-2080.
- Zubilewicz A, Hecquet C, Jeanny JC, Soubrane G, Courtois Y, et al. (2001) Two distinct signaling pathways are involved in FGF-2-stimulated proliferation of choriocapillary endothelial cells: A comparative study with VEGF. Oncogene 20: 1403-1413.
- Gama Sosa MA, De Gasperi R, Hof PR, Elder GA (2016) Fibroblast growth factor rescues brain endothelial cells lacking presenilin 1 from apoptotic cell death following serum starvation. Sci Rep 6: 30267.
- Chistiakov DA, Orekhov AN, Bobryshev YV (2015) Contribution of neovascularization and intraplaquehaemorrhage to atherosclerotic plaque progression and instability. ActaPhysiol (Oxf) 213: 539-553.
- Pai JK, Mukamal KJ, Rexrode KM, Rimm EB (2008) C-reactive protein (CRP) gene polymorphisms, CRP levels, and risk of incident coronary heart disease in two nested case-control studies. PLoS One 3: e1395.
- Dollard J, Kearney P, Clarke G, Moloney G, Cryan JF, et al. (2015) A prospective study of C-reactive protein as a state marker in Cardiac Syndrome X. Brain BehavImmun 43: 27-32.
- Bonaventura A, Mach F, Roth A, Lenglet S, Burger F, et al. (2016) Intraplaque expression of C-reactive protein predicts cardiovascular events in patients with severe atherosclerotic carotid artery stenosis. Mediators Inflamm 2016: 9153676.
- Gerner RR, Wieser V, Moschen AR, Tilg H (2013) Metabolic inflammation: role of cytokines in the crosstalk between adipose tissue and liver. Can J PhysiolPharmacol 91: 876-872.
- Bian F, Yang X, Zhou F, Wu PH, Xing S, et al. (2014) C-reactive protein promotes atherosclerosis by increasing LDL transcytosis across endothelial cells. Br J Pharmacol 171: 2671-2684.
- Hart SP, Alexander KM, MacCall SM, Dransfield I (2005) C-reactive protein does not opsonize early apoptotic human neutrophils, but binds only membrane-permeable late apoptotic cells and has no effect on their phagocytosis by macrophages. J Inflamm (Lond.) 2: 5.
- Eisenhardt SU, Habersberger J, Murphy A, Chen YC, Woollard KJ, et al. (2009) Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res 105: 128-137.
- Slevin M, Rovira N, Turu M, Luque A, Badimon L, et al. (2009) Modified C-reactive protein is expressed in adventitia and intimal neovessels from complicated regions of unstable carotid plaques. Open CircVasc J 2: 23-29.
- de la Torre R, Peña E, Vilahur G, Slevin M, Badimon L (2013) Monomerization of C-reactive protein requires glycoprotein IIb-IIIa activation: pentraxins and platelet deposition. J ThrombHaemost 11: 2048-2058.
- Slevin M, Matou-Nasri S, Turu M, Luque A, Rovira N, et al. (2010) Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol 20: 151-165.
- Peña E, de la Torre R, Arderiu G, Slevin M, Badimon L (2016) mCRP triggers angiogenesis by inducing F3 transcription and TF signaling in microvascular endothelial cells. ThrombHaemost 117: 357-370.
- Sternik L, Samee S, Schaff HV, Zehr KJ, Lerman LO, et al. (2002) C-reactive protein relaxes human vessels in vitro. ArteriosclerThrombVascBiol 22: 1865-1868.
- Boras E, Slevin M, Alexander MY, Aljohi A, Gilmore W, et al. (2014) Monomeric C-reactive protein and Notch-3 cooperatively increase angiogenesis through PI3K signaling pathway. Cytokine 69: 165-179.
- Ji SR, Ma L, Bai CJ, Shi JM, Li HY, et al. (2009) Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J 23: 1806-1816.
- Turu MM, Slevin M, Matou S, West D, Rodríguez C, et al. (2008) C-reactive protein exerts angiogenic effects on vascular endothelial cells and modulates associated signaling pathways and gene expression. BMC Cell Biol 9: 47.
- Slevin M, Matou S, Zeinolabediny Y,Corpas R, Weston R, et al. (2015) Monomeric C-reactive protein--a key molecule driving development of Alzheimer's disease associated with brain ischaemia? Sci Rep 5: 13281.
- Potempa LA, Yao ZY, Ji SR, Filep JG, Wu Y (2015) Solubilization and purification of recombinant modified C-reactive protein from inclusion bodies using reversible anhydride modification. Biophys Rep 1: 18-33.
- Sattar A, Rooney P, Kumar S, Pye D, West DC, et al. (1994) Application of angiogenic oligosaccharides of hyaluronan increases blood vessel numbers in rat skin. J Invest Dermatol 103: 576-579.
- Angulo J, Matou S (2007) Application of mathematical morphology to the quantification of in vitro endothelial cell organization into tubular-like structures. Cell MolBiol (Noisy-le-grand) 53: 22-35.
- Bigg HF, Rowan AD, Barker MD, Cawston TE (2007) Activity of matrix metalloproteinase-9 against native collagen types I and III. FEBS J 274: 1246-1255.
- Dol-Gleizes F, Delesque-Touchard N, Marès AM, Nestor AL, Schaeffer P, et al. (2013) A new synthetic FGF receptor antagonist inhibits arteriosclerosis in a mouse vein graft model and atherosclerosis in apolipoprotein E-deficient mice. PLoS One 8: e80027.
- Matou S, Colliec-Jouault S, Galy-Fauroux I, Ratiskol J, Sinquin C, et al. (2005) Effect of an oversulfatedexopolysaccharide on angiogenesis induced by fibroblast growth factor-2 or vascular endothelial growth factor in vitro. BiochemPharmacol 69: 751-759.
- Ornitz DM, Itoh N (2015) The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev DevBiol 4: 215-266.
- Xu C, Wang C, Cai QF, Zhang Q, Weng L, et al. (2015) Matrix Metalloproteinase 2 (MMP-2) Plays a Critical Role in the Softening of Common Carp Muscle during Chilled Storage by Degradation of Type I and V Collagens. J Agric Food Chem 63: 10948-10956.
- Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12: 9-18.
- Casar B, Arozarena I, Sanz-Moreno V, Pinto A, Agudo-Ibáñez L, et al. (2008) Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol Cell Biol 29: 1338-1353.
- Reis-Sobreiro M, Roué G, Moros A, Gajate C, de la Iglesia-Vicente J, et al. (2013) Lipid raft-mediated Akt signaling as a therapeutic target in mantle cell lymphoma. Blood Cancer J 3: e118.
- Matsumura N, Takami M, Okochi M, Wada-Kakuda S, Fujiwara H, et al. (2014) g-Secretase associated with lipid rafts. Multiple interactive pathways in the stepwise processing of ?-carbonyl-terminal fragment. J BiolChem 289: 5109-5121.
- Kang DE, Yoon IS, Repetto E, Busse T, Yermian N, et al. (2005) Presenilins mediate phosphatidylinositol 3-kinase/AKT and ERK activation via select signaling receptors. Selectivity of PS2 in platelet-derived growth factor signaling. J BiolChem 280: 31537-31547.
- Kim SK, Park HJ, Hong HS, Baik EJ, Jung MW, et al. (2006) ERK1/2 is an endogenous negative regulator of the gamma-secretase activity. FASEB J.; 20: 157-159.
- Wu JR, Yeh JL, Liou SF, Dai ZK, Wu BN, et al. (2016) Gamma-secretase inhibitor prevents proliferation and migration of ductusarteriosus smooth muscle cells through the Notch3-HES1/2/5 pathway. Int J BiolSci 12: 1063-1073.
- Wu J, Ye J, Zhu J, Xiao Z, He C, et al. (2016) Heparin-based coacervate of FGF-2 improves dermal regeneration by asserting a synergic role with cell proliferation and endogenous facilitated VEGF for cutaneous wound healing. Biomacromolecules 17: 2168-2177
- Pintucci G, Moscatelli D, Saponara F, Biernacki PR, Baumann FG, et al. (2002) Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. FASEB J 16: 598-600.
- Wang CH, Li SH, Weisel RD, Fedak PW, Dumont AS, et al. (2003) C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation 107: 1783-1790.
- Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. BiochimBiophysActa 1813: 1938-1945.
- Rieck PW, Cholidis S, Hartmann C (2001) Intracellular signaling pathway of FGF-2-modulated corneal endothelial cell migration during wound healing in vitro. Exp Eye Res 73: 639-650.
- Srinivasan R, Zabuawala T, Huang H, Zhang J, Gulati P, et al. (2009) Erk1 and Erk2 regulated endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS One 4: e8283.
- Chen J, Gu Z, Wu M, Yang Y, Zhang J, et al. (2016) C-reactive protein can upregulate VEGF expression to promote ADSC-induced angiogenesis by activating HIF-1α via CD64/PI3k/Akt and MAPK/ERK signaling pathways. Stem Cell Res Ther 7: 114.
- Adya R, Tan BK, Punn A, Chen J, Randeva HS (2008) Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signaling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res 78: 356-365.
- Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, et al. (2012) Downregulation of the Notch signaling pathway inhibits hepatocellular carcinoma cell invasion by inactivation of matrix metalloproteinase-2 and -9 and vascular endothelial growth factor. Oncol Rep 28: 874-882.
- Oh IS, Kim HG (2004) Effect of fibroblast growth factor-2 on migration and proteinases secretion of human umbilical vein endothelial cells. J MicrobiolBiotechnol 14: 379-384.
- Lin R, Liu J, Gan W, Yang G (2004) C-reactive protein-induced expression of CD40-CD40L and the effect of lovastatin and fenofibrate on it in human vascular endothelial cells. Biol Pharm Bull 27: 1537-1543.
- Cimmino G, Ragni M, Cirillo P, Petrillo G, Loffredo F, et al. (2013) C-reactive protein induces expression of matrix metalloproteinase-9: a possible link between inflammation and plaque rupture. Int J Cardiol 168: 981-986.
- Sluitjer JP, Pulskens WP, Schoneveld AH, Velema E, Strijder CF, et al. (2006) Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinase 8 and 9 with vulnerable carotid atherosclerotic lesions: a study in human endarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms. Stroke 37: 235-239.
- Wang Q, Stump R, McAvoy JW, Lovicu FJ (2009) MAPK/ERK1/2 and PI3-kinase signaling pathways are required for vitreous-induced lens fibre cell differentiation. Exp Eye Res 88: 293-306.
- Aimi F, Georgiopoulou S, Kalus I, Lehner F, Hegglin A, et al. (2015) Endothelial Rictor is crucial for midgestational development and sustained and extensive FGF-2-induced neovascularization in the adult. Sci Rep 5: 17705.