Research Article - (2023) Volume 10, Issue 2
Design and Evaluation of Vildagliptin Matrix Tablets by Response Surface Methodology
Deepika B* and
Sujatha K
Department of Pharmacy, Sri Ramachandra University, Chennai, India
*Correspondence:
Deepika B, Department of Pharmacy, Sri Ramachandra University, Chennai,
India,
Email:
Received: 07-Feb-2023, Manuscript No. IPADT-23-15667;
Editor assigned: 09-Feb-2023, Pre QC No. IPADT-23-15667 (PQ);
Reviewed: 23-Feb-2023, QC No. IPADT-23-15667;
Revised: 07-Mar-2023, Manuscript No. IPADT-23-15667 (R);
Published:
14-Mar-2023, DOI: 10.35841/2349-7211.10.2.11
Abstract
Currently, the use of plant based natural gums and mucilage are increased in manufacturing of pharmaceutical dosage forms as retarding polymers and excipients, because these natural plant based materials are biocompatible, biodegradable, free of side effects and economic. Therefore in the present research work, eight different formulations of vildagliptin matrix tablets were formulated with Hibisus leaves mucilage, zein powder and tamarind seed polysaccharide as natural polymers to sustain and extend the vildagliptin release from the matrix system and wet granulation technique used for preparation of matrix tablets. The prepared matrix tablets were evaluated for pre-compression and post-compression parameters. Hence the developed matrix tablets of all the formulations passed all the standard evaluation tests (pre-compression and post-compression parameters) and the difference between both the polymers in terms of drug release was examined. V1 formulation extended the release unto 24 hrs, considered as best formulation. The dissolution data of all formulations fitted into kinetic models like zero order equation, first order equation, Hixson Crowell equation, Higuchi equation and Korsemeyer-peppas equation in order to determine the order of drug release and mechanism. The release kinetics of best formulations showed zero order release and followed Higuchi mechanism.
Keywords
Vildagliptin; Tamarind seed polysaccharide; Zein powder; Matrix tablets; Sustained release
Introduction
Oral route is well known, important drug delivery system with
ease of administration and cost effective. In that sustained
release tablets are important drug delivery systems and are
designed to release the drug constantly and continuously for
prolonged period of time in order to maintain the drug
plasma concentration within the therapeutic level as
compared to other dosage forms. Diabetes Mellitus (DM)
belongs to a group of metabolic diseases, which are
characterized by prolonged maintenance of high blood sugar
levels, which are divided into two major groups: Type-1
diabetes and type-2 diabetes. Type-1 is caused by pancreatic
β-cell dysfunction. Vildagliptin is an amino acid derivative,
which stimulates insulin secretion from pancreas, and
decreases the plasma (blood) glucose levels. The therapeutic
action of vildagliptin is depends upon functioning beta cells in
the pancreatic islets [1].
The objective of the study aimed to develop and evaluate
vildagliptin sustained release matrix tablet using zein powder
and tamarind seed polysaccharide at different concentrations.
Zein powder (prolamine family) is rate retarding polymer, composed of high amounts of hydrophobic amino acids
(>50%) like proline, glutamine and asparagines. Tamarind
seed polysaccharide (Tamarindus indica) is also retarding
agent belongs to leguminous tree (Fabacea family) bearing
edible fruit [2].
Materials and Methods
Materials
Vildagliptin is an antidiabetic drug, was gifted from Shodhana
laboratories Ltd, Hyderabad, India. Zein and tamarind seed
polysaccharide were collected from Cisco research labs pvt
ltd, Mumbai. Microcrystalline cellulose (Avicel PH 101) was a
gift from Signet chemicals Mumbai, India. Isopropyl alcohol
was gift from Anshul agencies, Mumbai, India [3]. Magnesium
stearate and talc were procured from S.D. fine chemicals,
Mumbai, India. All the chemicals used in the study were of
analytical grade.
Extraction of Mucilage from Hibiscus rosa-sinensis Leaves
Fresh leaves of Hibiscus rosa-sinensis (Class: Magnoliopsida;
Family: Malvaceae) were used for mucilage extraction. Dirt
and debris associated with collected leaves were removed by
cleaning repeatedly with adequate water [4]. The cleaned
leaves were subjected to drying until they become crispy. The
dried leaves were crushed and maintained under soaking for
5-6 hours. All soaked leaves were boiled for 30 min and later
on kept undisturbed for 1 hour to allow the complete release
of mucilage into water. An eight-fold muslin cloth bag was
used to extract the mucilage by removing marc from solution.
Then resultant filtrate solution was mixed with acetone (three
times to filtrate’s volume) to precipitate the mucilage. The
precipitated mucilage was separated carefully and dried in an
oven at 40°C. The dried product was ground to get HRLM
powder, passed through sieve number # 80 and stored in
desiccator at 35°C and 45% relative humidity till use [5].
Extraction of Tamarind Seed Polysaccharide (TSP)
Seeds from tamarind fruits (Plant: Tamarindus indica L.; sub-family:
Caesalpinioideae; Family: Fabaceae/Leguminosae)
were collected from local area and washed with distilled
water to remove adherent material. Air dried seeds were
roasted at 140°C for 10 min to enable the formation of a gap
between testa and kernel in each seed [6]. Then roasted seeds
were hammered to break the brittle testa and kernel was
separated from each seed. All the separated kernels were
cleaned with water, air dried and powdered to get Tamarind
Kernel Powder (TKP).
A slurry was prepared by mixing 20 g of TKP with 200 ml
distilled water. The resultant slurry was added to 800 ml water
and boiled for 20 min with continuous stirring. Then obtained
thin clear solution was cooled and kept undisturbed overnight
to enable the maximum settling out of proteins and fibers. A
clear supernatant liquid (formed after the centrifugation at
5000 rpm for 20 min) was separated from solution and mixed
with doubled volume proportion of absolute ethanol to
facilitate TSP precipitation [7]. The precipitate was then
collected, washed with absolute ethanol and dried in an oven
for 10°C hours at 50°C. Completely dried TSP was powdered,
passed through sieve number 60 and stored in a desiccator
until further use.
Preparation of SR Matrix Tablets of Miglitol
Formulation compositions were developed using suitable
experimental design to study the effect of selected three
factors on predetermined response. Pre-formulation studies
like drug-excipient compatibility study and flow property
evaluation were conducted to know the suitability of
developed blend compositions for tablet compression (Table
1).
Experimental design: Total eight tablet formulations were
developed for each drug by applying 23 factorial design using
‘Design expert software’. The objective of this design was to
study the effect of three independent factors like amount of
HRLM (A), amount of ZP (B) and amount of TSP (C) on drug
release characteristics. Each factor was studied at two levels
designated as -1 (60 mg; 15% w/w) and +1 (80 mg; 20% w/w).
The randomized combinations were generated based on
Design of Experiments (DOE). The designed combinations of
factors were used as a base to develop eight different
formulation compositions for each drug [8]. The response
(dependent) variable studied for each formulation was
‘cumulative % drug release after 24 hours.
| Formulation code |
Level of factor in formulations |
| A (Amount of HRLM) |
B (Amount of ZP) |
C (Amount of TSP) |
| F1 |
-1 |
-1 |
-1 |
| F2 |
1 |
-1 |
-1 |
| F3 |
-1 |
1 |
-1 |
| F4 |
1 |
1 |
-1 |
| F5 |
-1 |
-1 |
1 |
| F6 |
1 |
-1 |
1 |
| F7 |
-1 |
1 |
1 |
| F8 |
1 |
1 |
| -1 → 60 mg; +1 → 80 mg |
Table 1: Randomized possible compositions as per DOE.
Pre Formulation Studies
Physicochemical properties of the drug and its blend with
other excipients can be investigated by conducting pre
formulation studies. The formulation blends prepared as per
above developed compositions were used for:
• Compatibility study.
• Evaluation of flow properties.
Standard graph of drug was also constructed in pre
formulation studies.
Compatibility Study
Blended samples were analyzed using Fourier Transform
Infrared (FTIR) spectrophotometer to interpret the
interactions of drug with polymers and other ingredients. The
powder blend along with potassium bromide was used for
FTIR studies. The IR spectrum of a) Pure drug; b) Physical
mixture containing drug, polymers and other ingredients,
were taken, interpreted and compared with each other [9].
Characterization of Formulation Powder Mixture for
Flow Property
The physicochemical properties of a formulation blend are
deciding factors of tablet compression and output quality.
Mainly, flow property of blend (powder/granular) is an
important characteristic that plays a major role in
compression process. The following properties of formulation
blend were determined to interpret its flow property [10].
Angle of Repose
It is defined as maximum possible angle between surface of
blend’s pile and its horizontal plane. If more powder bed is
added to previously formed pile, it slides down the sides until
mutual friction of particles producing a surface angle θ is in
equilibrium with gravitational force. The angle of repose (θ)
values of formulation blends were measured using fixed
funnel [11]. A funnel was tightly fixed to a stand by keeping
funnel’s lower tip at predetermined known height (h) above a
graph paper placed on a flat horizontal surface. Pouring of
formulation blend through the funnel was started. It was
continued until formed apex of conical pile just touches the
lower tip of funnel stem. Then, radius (r) of the base of conical
pile was measured (Table 2). The angle of repose (θ) was
calculated using below mentioned formula:
θ=Tan-1[h/r]
| Flow property |
Angle of repose (degrees) |
| Excellent |
25–30 |
| Good |
31–35 |
| Fair—aid not needed |
36–40 |
| Passable—may hang up |
41–45 |
| Poor—must agitate, vibrate |
46–55 |
| Very poor |
56–65 |
| Very, very poor |
>66 |
Table 2: Pharmacopoeial specifications for angle of repose.
Compressibility Index and Hausner Ratio
The compressibility index (Carr’s Index) is a measure for flow
nature of a formulation blend to be compressed. It is
calculated using both bulk and tapped density values of
powder blend.
Bulk density: Bulk density (ρb) value can be calculated by
dividing the mass of powder blend with its bulk volume and is
expressed as g/ml. Its value primarily depends on powder
particulate properties like size distribution, shape and the
tendency of particles to adhere together [12]. The dried 10 ml
cylinder was taken and a fixed Mass (M) of blend was
introduced into it. Inside content was carefully leveled
without compacting and noted the value of unsettled
apparent Volume (V0). The bulk density was calculated using
following formula:
ρb = M/V0
Tapped density: This value (ρtap) is represented by the ratio of
blend’s total mass (M) to its final tapped Volume (Vf). First,
the tapping of powder blend was done for 500 times and its
volume was measured. Then tapings were increased up to
750 times and the volume was noted for second time. Tapping
was continued until the difference between two consecutive
volumes was <2%. Then Vf was measured to the nearest
graduated unit. The tapped density was calculated in g/ml
using below mentioned formula:
ρtap = M/Vf
Carr’s Index (CI) and Hausner ratio are the measures of inter
particulate interactions in a powder blend (Table 3). The
values of bulk and tapped densities will be closer in a free
flowing powder due to les significant interactions. CI and
Hausner ratio were calculated using the following formula:
Carr’s index=((ρtap–ρb)/ρtap) X 100; and Hausner ratio=ρtap/ρb
| Compressibility index (%) |
Flow character |
Hausner ratio |
| ≤ 10 |
Excellent |
1.00–1.11 |
| 11-15 |
Good |
1.12–1.18 |
| 16-20 |
Fair |
1.19–1.25 |
| 21-25 |
Passable |
1.26–1.34 |
| 26-31 |
Poor |
1.35–1.45 |
| 32-37 |
Very poor |
1.46–1.59 |
| >38 |
Very, very poor |
>1.60 |
Table 3: Pharmacopoeial specifications for Carr’s index and Hausner ratio.
Construct on of Standard Graph of Drug(s)
Calibration curves of three drugs were plotted separately. Curve
of each drug was drawn in 0.1 N HCl and pH 6.8 phosphate
buffers, separately (Table 4). Each drug was scanned
spectrophotometrically at its respective λ max value as
follows:
| Drug |
λmax |
| Miglitol |
220 nm |
| Nateglinide |
210 nm |
| Vildagliptin |
210 nm |
Table 4: Absorption maxima of drug.
Preparation of 0.1 N HCl: 8.5 ml of concentrated HCl was
taken in 1000 ml volumetric flask and diluted with distilled
water up to mark.
Preparation of pH 6.8 phosphate buffer: It was prepared by
mixing 250 ml of 0.2 M potassium dihydrogen phosphate
solution with 112 ml of 0.2 M sodium hydroxide in a 1000 ml
volumetric flask. The resultant mixture was diluted with
distilled water up to mark.
Preparation of standard solution samples:
• First stock solution was prepared by dissolving 100 mg
drug in 100 ml of concerned medium (0.1 N HCl or pH 6.8
phosphate buffer), so as to get 1000 μg/ml concentration.
• Then a sample of 10 ml from above stock was diluted to
100 ml using same medium, so as to geta second solution
with 100 μg/ml concentration.
• Accurately measured aliquot portions of second solution,
like 0.2 ml, 0.4 ml, 0.6 ml, 0.8 ml, 1.0 ml and 1.2 ml were
transferred in to 10 ml volumetric flasks. Then all the
samples were diluted up to mark with medium. The final
concentrations of diluted solutions were ranged from 2
μg/ml-12 μg/ml.
• Absorbance of each diluted standard drug solution sample
was scanned in UV-Visible spectrophotometer at its
respective λ max using same medium as the blank. A
linear graph was plotted by taking drug concentration on
X-axis and absorbance on Y-axis and the values of slope
(m) and Y-intercept were noted.
Formulat on of SR Matrix Tablets
Matrix tablets of each drug were prepared by direct
compression method. Drug and selected polymer powders
were passed through sieve number 80 meshes to get uniform
fineness. Other excipients were screened through 60 mesh
sieve. Formulation bulk was developed using Micro Crystalline
Cellulose (MCC) as filler. Magnesium stearate and talc were
used to lubricate the formulation blends developed as per
experimental design. Lubricated blends were compressed using 9 mm flat faced punches on eight station rotary
compression machine (Cadmach Machinery Pvt. Ltd.,
Ahmedabad).
Evaluation of Compressed Tablets (Post-Formulation
Studies)
Compressed tablets were characterized for their
physicochemical properties by conducting post-compression
studies. First, all the formulations were evaluated by in vitro methods. The results of in vitro studies were analyzed,
interpreted and selected best optimized formulation(s) for
each drug [13]. The stability of selected tablet batches was
studied and then best formulations were evaluated by in vivo methods. Tablets in each formulation were evaluated for
weight variation, thickness, hardness, friability, content
uniformity, swelling index and drug release characteristics
during in vitro studies.
Weight Variation Test
Twenty tablets from each formulation batch were selected
randomly and noted their individual weights (n=6). The
average weight of one tablet was estimated and calculated
percentage deviation of individual reading from average value
using following formula:
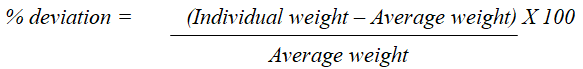
The % deviation allowed for 400 mg (average weight) tablets
as per pharmacopoeia is 5%. The number of tablets
possessing more than ± 5% deviation were identified and
noted, if any. Mean weight of tablets in each batch was
calculated and represented with Standard Deviation (SD)
values (Table 5).
| Average weight of tablets (mg) (I.P) |
Average weight of tablets (mg) (U.S.P) |
Maximum percentage deviation allowed |
| Less than 80 |
Less than 130 |
10 |
| 80–250 |
130-324 |
7.5 |
| More than 250 |
More than 324 |
5 |
Table 5: Pharmacopoeial specifications for tablet weight variation.
Thickness
Tablet thickness is an important characteristic that ensures
reproducing appearance twenty tablets were taken randomly
from each formulation and noted their thickness values using
digital Vernier calipers (n=6). Thickness of the tablets should
be controlled in ± 5%. So verified and noted the deviated
tablets in each batch. Mean value of each formulation were
calculated and represented with SD values.
Hardness
The force applied along tablet diameter is considered as its
hardness which is crucial to show resistance towards chipping,
abrasion or breakage under conditions of storage,
transportation and handling before usage. The hardness of 20
tablets from each batch was noted using Monsanto hardness
tester (n=6) and mean values with SD were presented.
Friability
Mechanical strength of prepared dosage units can be
estimated by measuring percentage friability using Roche
friabilator which consists of one plastic chamber that is set to
revolve 25 rpm dropping the tablets from 6 inches’ height
with each revolution. The collective weight (Wo) of 20 tablets
from each formulation was noted (n=6) and pre-weighed
dosage forms were placed in friabilator to revolve for 4
minutes [14]. Then all the tablets were re-weighed collectively
and noted as W. The difference in two weights is a measure of
friability and it can be expressed in percentage as:
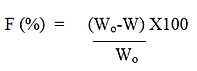
Where;
Wo=Weight of the tablets before test.
W=Weight of the tablets after test.
Mean friability percentage was calculated for each batch and
represented with corresponding SD value. The % friability
should lie in range of 0.5%-1% as per pharmacopoeia.
Content Uniformity
Randomly selected 20 tablets from each formulation were
weighed and one tablet average weight was calculated. Then
all 20 tablets were powdered and the powder equivalent to
one average tablet weight was taken. The weighed tablet
powder was allowed to dissolve in 250 ml of pH 6.8 phosphate
buffer on a rotary shaker overnight. The resultant solution was
centrifuged to get supernatant. The absorbance of
supernatant (after required dilution) was measured using a
UV-visible spectrophotometer at required λmax of drug.
Swelling Study
The swelling behavior of all prepared tablets was determined
by estimating their Swelling Index (SI) value. SI indicates the
extent of swelling in terms of percent weight gain by tablet.
Three tablets form each formulation (n=3) were selected and noted their initial weights (W0). Pre-weighed tablets were
placed in 8 separate petri dishes (formulation-wise) each
containing 20 ml of pH 6.8 phosphate buffer. All the tablets
were removed from petri dish after 1 hour (t=1) and placed
on a tissue paper for 5 minutes. Then weights of tablets (Wt)
were noted and again paced in same petri dishes. In the same
way, Wt values were noted after another one hour (t=2) and
then later on done for every 2 hours up to 12 hours. The SI
value for all tablets was calculated using following formula:
SI=((Wt-W0)/W0) × 100
Where;
Wt=Weight of a tablet at ‘t’ time.
W0=Initial weight of a tablet at ‘0’ time.
Drug Release Study
Multiple dissolution apparatus (Electro lab India pvt. ltd.) was
used for in vitro drug release study. Dissolution was carried
out using USP type-I (basket) method in 900 ml of medium for
24 hours. The medium used was 0.1 N HCl during first 2 hours
and then replaced with pH 6.8 phosphate buffers for
continuing remaining study. Samples (5 ml) were withdrawn
at predetermined time points and replaced with equal volume
of same fresh medium at each collection. The collected
samples were scanned in UV-visible spectrophotometer at
drug specific λmax. The cumulative % drug release after each
collected time point was calculated using standard graph of
drug [15].
Drug Release Kinetic Studies
Drug release data obtained through dissolution study was
fitted into various kinetic models to assess the kinetics and
mechanism of drug release. The relevant linear plots were
drawn using MS-Excel-2010 and generated regression
equations for each plot. The extent of linearity was assessed
with regression coefficient (r2) value. The model which results
a plot with highest linearity was selected as best fit model for
fitted drug release data. The kinetic models and their
equations used in data fitting were as follows:
• Zero order model (Qt=Qo–Kot): Cumulative % drug
released versus time plot was drawn.
• First order model (ln Q=ln Qo–K1t): Log cumulative %
drug remaining to be released versus time plot was drawn.
• Higuchi model (Q=Kht1/2): Cumulative % drug released
versus square root of time plot was drawn.
• Korsmeyer-Peppas model (Mt/Mα=Ktn): Log cumulative %
drug released versus log time plot was drawn.
• Hixson-Crowell cubic root law model (Qo1/3–Qt1/3=Kt):
Cubic root of cumulative % drug remaining to be released
versus time plot was drawn.
In above kinetic equations,
• Qi and Ki=Stand for amount of drug release and release
constant, respectively.
• Mt/Mα=Represents fractional drug release.
• ‘t’=Stands for time point of concerned.
• ‘n’=Diffusional release exponent.
Two-Level Factorial Optimization and Mathematical
Modeling
A randomized two level full factorial design was used to
analyze the obtained data. After obtaining post-experimental
results (in vitro drug release data), a multiple regression
analysis was applied to evaluate the regression coefficients of
generated mathematical model. The following statistical
model incorporating linear terms and interactive terms of
three factors studied was used to test the responses:
Y= β0+β1A+ β2B+ β3C+ β4AB+ β5AC+ β6BC+ β7ABC
Where;
Y=Response variable.
β0=Arithmetic mean response of all eight runs.
β1, β2 and β3=Estimated coefficients for individual factors A, B
and C respectively.
β4, β5, β6 and β7=Estimated coefficients for interaction terms
AB, AC, BC and ABC respectively.
A, B and C=Indicate main effects that represent the average
result of changing one factor at a time from its low to high
value.
AB, AC and BC are the interaction terms that indicate possible
changes in response when two factors are altered together.
ABC is an interactive term which represents the manner how
response changes when all three factors together subjected
to alteration.
The significance of each term in above said statistical model
was evaluated by applying ANOVA test. Only, the significant
terms were included in final model. The effect of investigated
factors on dependent variable was visualized by drawing ‘3D
surface and contour plots’. The optimized formulation and
its predicted response were suggested by used statistical
model. Then suggested optimized formulation was confirmed
as the best by comparing its practically observed drug
release with predicted response.
In vitro Drug Release: Marketed Versus Optimized
Formulation
Dissolution study was conducted for a marketed product of
concerned drug using same parameters and procedure as that
used for developed formulations. The drug release profile of
selected best optimized formulation was compared with that
of marketed tablet.
Stability Studies
Best formulations were selected based on the results of in
vitro studies. Accelerated stability studies were conducted for
selected best optimized formulation(s) using stability
chamber (Bio-Techniques equipment Ltd.). Tablets of each
best formulation were divided into 3 batches; strip packed and stored at three different conditions as per ICH guidelines.
The three storage conditions used in these studies were as
follows:
• Room temperature.
• 30°C temperature and 60% Relative Humidity (RH).
• 40°C temperature and 75% Relative Humidity (RH).
Dosage form samples from each batch were collected at
predetermined time intervals of 0, 30, 60, 90 and 180 days.
Collected sample units were evaluated for physicochemical
parameters like appearance, weight variation, thickness,
hardness, friability, drug content and drug release.
In vivo Studies
In vivo studies were conducted to estimate the pharmacokinetic
parameters of drug to support in vitro drug release results.
These animal studies were performed in male New Zealand
white rabbits weighing 2.0 kg to 2.5 kg, after getting approval
from Institutional Animal Ethical Committee (IAEC) (approval no.
IAEC/05/NCOP/2019). All the rabbits were procured from a
registered vendor. Rabbits were divided into 3 groups each
consisting of 3 animals and fasted overnight, allowed to take
drinking water alone before experimentation. First group
(reference) was administered with drug solution (prepared by
dissolving dose equivalent quantity in sufficient distilled water). Second and third groups
received marketed product and selected best formulation,
respectively. All the dose administrations to rabbits were
done via gastric intubation. Blood sampling was done from
ear vein by holding the animals in rabbit restrainers. Blood
samples were collected into heparinized tubes at
predetermined intervals of 0.5, 1, 1.5, 2, 4, 8, 12 and 24 hours
post administration. Collected blood samples were subjected
to cooling centrifugation (Heraeus Biofuge Fresco centrifuge,
Germany) at 3500 × g of Relative Centrifugal Force (RCF) and
4°C for 5 minutes to obtain the plasma. Separated test plasma
samples were stored at -70°C until further analysis [16].
Analysis of Plasma Drug Concentration using HPLC
A sensitive Reverse Phase High Performance Liquid
Chromatography (RP-HPLC) method (Shimadzu corporation,
Japan) was used for the estimation of drug in plasma. The
samples were analyzed using RP C18 column (250 mm x 4.6
mm, 5 μm, Phenomenex Luna) as stationary phase. The
sample run time for analysis was 10 min. Lab solutions
software was used to perform all analysis operations and
interpret the analyzed data. The following HPLC parameters
were used in analysis of samples (Table 6).
| HPLC parameter |
Vildagliptin |
| Mobile phase |
pH 4.6 phosphate buffer and acetonitrile in the ratio of 60:40 v/v |
| Flow rate |
1 ml/min |
| λmax |
210 nm |
| Internal standard |
Metformin |
| First, calibration curve of drug in rabbit plasma was plotted using above said HPLC parameters. |
Table 6: HPLC Parameters for the analysis of drugs.
Construction of Calibration Curve for Drug
Preparation of stock and working standard solutions: First, a
stock solution (100000 ng/ml) was prepared by dissolving
accurately weighed amount (10 mg) of drug in 100 ml of HPLC
grade methanol. Then 1 ml of above solution was diluted 100
times to get second stock (1000 ng/ml) using same grade
methanol. Further dilutions were made to the second stock
solution to get working standard solutions in a concentration
range of 200 ng/ml-1000 ng/ml. Five standard drug solutions
with concentration of 200 ng/ml, 400 ng/ml, 600 ng/ml, 800
ng/ml and 1000 ng/ml were used for standard curve.
Preparation of Internal Standard solution (IS): Accurately
weighed internal standard drug (10 mg) was dissolved in 10ml
of HPLC grade methanol to get a stock solution (1000 μg/ml).
Then a working internal standard solution (10 μg/ml) (to be
used in further analysis) was prepared by making dilutions to
previously obtained stock solution. Preparation of HPLC
samples for Standard graph: Blood samples (drug free) were
collected from healthy male rabbits and centrifuged to get
plasma. Standard drug solutions (each 100 μl) were placed in
separate polypropylene centrifuge tubes and 100 μl of plasma
sample was added to each. Then internal standard solution
(100 μl) was added to each tube and shaken for 1 min. Each
resultant mixture was allowed to precipitate by adding
methanol (100 μl) and overtaxing for 1 min. Then all the tubes
were subjected to centrifugation for 20 min at 3000 rpm to
get clear supernatants. Then separated clear supernatants
were used as HPLC samples for construction of standard
graph. Each sample aliquot (20 μl) with known drug
concentration was injected into HPLC column and analyzed at
specified wavelength. The peak area of both drug as well as
internal standard was noted and peak area ratio was
calculated for each sample. Then concentration versus peak
area ratio plot (calibration curve) was drawn using MS-Excel.
Peak area ratio was calculated as follows:
Peak area ratio=peak area of drug/peak area of internal
standard.
Analysis of Test Plasma Samples
Test plasma sample and internal standard (each 100 μl) were
mixed in a centrifuge tube. This mixture was shaken for 1 min.
To this mixture, 100 μl of methanol was added for
precipitation. The resultant was vortexed and centrifuged for
20 min at 3000 rpm. Then the supernatant was collected and
filtered. Now, the collected filtrate was considered as HPLC
test plasma sample. Then 20 μl of test sample was injected in
to HPLC system for analysis. All test plasma samples were
analyzed using same HPLC parameters and obtained ‘peak
area ratio’ values. The respective drug concentrations for
resulted ‘peak area ratio’ values were calculated using
calibration curve of drug. The obtained data of plasma drug
concentrations against different time points was applied in
Kinetical TM software to calculate important pharmacokinetic
parameters like peak plasma concentration (Cmax), Time
required to attain Cmax (tmax), Area Under Curve (AUC)
and Mean Residence Time (MRT) (Table 7).
| Ingredients (mg) |
Formulation code |
| V1 |
V2 |
V3 |
V4 |
V5 |
V6 |
V7 |
V8 |
| Vildagliptin |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
| HRLM |
60 |
80 |
60 |
80 |
60 |
80 |
60 |
80 |
| ZP |
60 |
60 |
80 |
80 |
60 |
60 |
80 |
80 |
| TSP |
60 |
60 |
60 |
60 |
80 |
80 |
80 |
80 |
| MCC |
162 |
142 |
142 |
122 |
142 |
122 |
122 |
102 |
| Magnesium stearate |
4 |
4 |
4 |
4 |
4 |
4 |
4 |
4 |
| Talc |
4 |
4 |
4 |
4 |
4 |
4 |
4 |
4 |
| Total weight |
400 |
400 |
400 |
400 |
400 |
400 |
400 |
400 |
Table 7: Formulation compositions for vildagliptin SR matrix tablets.
Results and Discussion
Vildagliptin SR Matrix Tablets
Preformulation Studies: Compatibility Study:
The FTIR analysis of pure vildagliptin powder has resulted a
spectrum with following peaks related to specific stretching’s
(Figure 1).

Figure 1: FTIR spectrum of pure vildagliptin.
The FTIR spectrum of powder mixture containing drug and
excipients has retained all the above identified peaks related
to pure vildagliptin. So, the existence of compatibility among
drug and other excipients in a formulation blend was
confirmed (Figure 2 and Table 8).

Figure 2: FTIR spectrum of mixture containing vildagliptin
+other ingredients.
| Wave number of peak formation (cm-1) |
Related stretching |
| 2220.14 |
-C≡N |
| 1647.98 |
>C=O |
| 1606.24 |
>N-H |
| 1271.23 |
=C-N |
| 1045.01 |
>C-N (ring) |
| 3743.55 |
-O-H |
Table 8: Peak regions in FTIR spectrum of Vildagliptin.
Characterization of Formulation Powder Mixture for its
Flow Property
All the values of angle of repose, Carr’s index and Hausner
ratio were bound to Pharmacopoeial limits and these results
have given an idea about flow nature of all developed
formulation blends. All the formulation powder mixtures
have possessed a flow characteristic of Excellent too good.
So, all blends were proved to be suitable for direct
compression (Table 9).
| Formulation blend |
Angle of repose (in°) |
Carr’s index (%) |
Hausner ratio |
| V1 |
23.82 |
12.73 |
1.22 |
| V2 |
24.29 |
11.28 |
1.31 |
| V3 |
23.88 |
10.76 |
1.07 |
| V4 |
20.79 |
11.49 |
1.13 |
| V5 |
23.26 |
12.29 |
1.11 |
| V6 |
24.66 |
12.17 |
1.12 |
| V7 |
21.61 |
10.09 |
1.13 |
| V8 |
23.19 |
11.37 |
1.04 |
Table 9: Flow property characterization of vildagliptin formulation blends.
Standard Graph of Vildagliptin
Two standard graphs of vildagliptin plotted in different media
have shown a good linearity with R2 value of 0.9996 (0.1N HCl)
and 0.9997 (pH 6.8 phosphate buffer) (Table 10). The obtained
absorbances were proportional to concentrations. Both
graphs were in accordance with ‘Beer-Lambert’ law (Figures 3
and 4).
| Concentration (µg/ml) |
Absorbance (0.1 N HCl) |
Absorbance (pH 6.8 phosphate buffer) |
| 0 |
0 |
0 |
| 2 |
0.113 |
0.161 |
| 4 |
0.206 |
0.296 |
| 6 |
0.311 |
0.441 |
| 8 |
0.421 |
0.582 |
| 10 |
0.518 |
0.729 |
| 12 |
0.619 |
0.869 |
Table 10: Standard graph of Vildagliptin using UV method.

Figure 3: Standard graph of vildagliptin in 0.1 N HCl using UV
method.

Figure 4: Standard graph of vildagliptin in pH 6.8 phosphate buffer
using UV method.
Evaluation of Compressed Tablets (Post-Formulation
Studies
In vitro studies
Weight variation, hardness, thickness: The weights of
individual tablets in each formulation were uniform and
not deviated from 398.78 ± 1.64 mg (V6) to 403.71 ±
0.99 mg (V5). The hardness values of all prepared tablets
were ranged from 4.1 ± 0.13 kg/cm2 (V3) to 4.3 ± 0.16 kg/cm2 (V5). The thickness of all tablets was uniform and no
deviation has been noticed as per standard pharmacopoeial
values as per pharmacopoeial limits. The average weights of
eight formulations were ranged (Table 11).
| Formulation |
Weight (mg) |
Hardness (kg/cm2) |
Thickness (mm) |
| V1 |
402.46 ± 1.27 |
4.2 ± 0.33 |
3.47 ± 0.21 |
| V2 |
401.74 ± 0.91 |
4.2 ± 0.19 |
3.49 ± 0.31 |
| V3 |
403.27 ± 1.49 |
4.1 ± 0.13 |
3.46 ± 0.29 |
| V4 |
400.66 ± 1.34 |
4.2 ± 0.27 |
3.48 ± 0.09 |
| V5 |
403.71 ± 0.99 |
4.3 ± 0.16 |
3.47 ± 0.11 |
| V6 |
398.78 ± 1.64 |
4.2 ± 0.22 |
3.49 ± 0.17 |
| V7 |
399.56 ± 1.42 |
4.2 ± 0.48 |
3.51 ± 0.07 |
| V8 |
402.09 ± 0.81 |
4.1 ± 0.15 |
3.48 ± 0.45 |
| *All the values are represented as Mean ± SD |
Table 11: Mean weight, hardness and thickness of Vildagliptin formulation.
Friability and content uniformity: All resulted values of
percentage friability were within the pharmacopoeial
standard range (0.5%-1%). The friability values of formulations
were ranged from 0.52 ± 0.13% (V7) to 0.72 ± 0.11% (V4). The
% drug content (assay) values in all formulations were uniform
and maximum to prescribed dose. These values were ranged
from 98.34 ± 0.55% (V8) to 102.46± 2.01% (V4). The
obtained results were found to be acceptable as per
pharmacopoeial standards (Table 12).
| Formulation |
Friability (%) |
Drug content (%) |
| V1 |
0.69 ± 0.14 |
101.06 ± 1.03 |
| V2 |
0.71 ± 0.05 |
99.41 ± 0.84 |
| V3 |
0.67 ± 0.18 |
98.79 ± 1.21 |
| V4 |
0.72 ± 0.11 |
102.46 ± 2.01 |
| V5 |
0.58 ± 0.26 |
99.38 ± 0.67 |
| V6 |
0.66 ± 0.14 |
99.18 ± 0.79 |
| V7 |
0.52 ± 0.13 |
101.45 ± 1.49 |
| V8 |
0.67 ± 0.12 |
98.34 ± 0.55 |
| *All the values are represented as Mean SD |
Table 12: Percentage friability and drug content of vildagliptin formulations.
Swelling index: From the results Table 1 and Figure 1 of
swelling study, it was understood that all used polymers in
vildagliptin formulations have a property of hydrophilicity
which is responsible for tablet swelling. The enhancement in
swelling percentagehasbeen increased with time up to 12
hours. The hike in SI values of formulations has been noticed
with enhanced polymer proportion within a tablet dosage
form. Comparatively, TSP polymer was found to be more
hydrophilic with enhanced swelling nature and high matrix
forming ability [17].
Formulation N1 having a composition of three polymers each
at lower amount (60 mg) has shown less SI values, whereas
more swelling had been seen with N8 tablets due to
higher quantity (80 mg) of each polymer (Table 13 and Figure
5).
| Time (hrs) |
Swelling index (%) |
| V1 |
V2 |
V3 |
V4 |
V5 |
V6 |
V7 |
V8 |
| 0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| 1 |
16.26 ± 1.22 |
14.59 ± 1.82 |
17.94 ± 1.19 |
15.62 ± 1.46 |
18.34 ± 1.97 |
20.37 ± 1.07 |
16.94 ± 1.33 |
30.51 ± 1.89 |
| 2 |
42.94 ± 1.37 |
36.61 ± 1.54 |
36.84 ± 1.37 |
37.42 ± 2.52 |
42.64 ± 1.75 |
36.94 ± 1.55 |
39.64 ± 1.84 |
51.77 ± 2.87 |
| 4 |
47.85 ± 1.46 |
61.27 ± 1.07 |
54.87 ± 2.66 |
66.09 ± 2.77 |
63.84 ± 2.66 |
81.44 ± 2.67 |
69.71 ± 2.67 |
92.64 ± 3.64 |
| 6 |
88.34 ± 2.03 |
92.48 ± 2.64 |
88.21 ± 2.49 |
97.18 ± 1.85 |
102.45 ± 3.27 |
124.61 ± 2.97 |
102.64 ± 3.82 |
133.52 ± 4.28 |
| 8 |
107.84 ± 2.74 |
114.027 ± 2.97 |
116.94 ± 3.16 |
125.67 ± 3.49 |
127.64 ± 3.86 |
143.67 ± 3.22 |
131.27 ± 4.67 |
161.94 ± 4.97 |
| 10 |
144.87 ± 2.67 |
152.94 ± 3.45 |
166.26 ± 3.78 |
162.42 ± 4.09 |
156.94 ± 4.84 |
187.61 ± 4.67 |
171.55 ± 4.98 |
194.76 ± 5.23 |
| 12 |
182.36 ± 3.29 |
203.48 ± 3.66 |
195.62 ± 3.87 |
234.15 ± 4.86 |
221.64 ± 4.67 |
262.34 ± 4.99 |
243.27 ± 4.67 |
273.45 ± 5.87 |
Table 13: Swelling index of vildagliptin SR matrix tablets.

Figure 5: Swelling behavior of vildagliptin SR matrix tablets.
Drug release study: The observed release pattern from tablets
in dissolution study was in sustained manner. No considerable vildagliptin release was seen during first two hours in 0.1N
HCL medium. Then a gradual hike in drug release was
noticed in pH 6.8 phosphate buffer during 2-24 hours. The
results revealed that the extent of drug release was affected
by both type and amount of rate retarding polymer. The
quantity of each polymer in every formulation has shown
its effect on drug release. A reduced drug release had
been noticed with increased polymer quantity and the
more impact was shown by TSP followed by HRLM and ZP.
Maximum drug release (93.63 ± 2.66%) has been noticed
from V1 formulation because of having a composition
of three polymers each at lower level (60 mg). The drug
release from V8 formulation was minimum (74.35 ± 1.87%)
due to more swelling and matrix formation by three
retardant polymers each at higher level (80 mg) (Table 14 and Figure 6).
| Time (hrs) |
Cumulative % of vildagliptin release |
| V1 |
V2 |
V3 |
V4 |
V5 |
V6 |
V7 |
V8 |
| 0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| 0.5 |
0.21 ± 0.05 |
0.38 ± 0.08 |
0.21 ± 0.07 |
0.21 ± 0.03 |
0.38 ± 0.06 |
0.03 ± 0.07 |
0.21 ± 0.08 |
0.38 ± 0.06 |
| 1 |
0.73 ± 0.14 |
0.56 ± 0.19 |
0.56 ± 0.17 |
0.38 ± 0.13 |
0.56 ± 0.14 |
0.56 ± 0.16 |
0.38 ± 0.14 |
0.56 ± 0.12 |
| 1.5 |
1.08 ± 0.19 |
0.91 ± 0.21 |
1.26 ± 0.24 |
0.91 ± 0.18 |
1.08 ± 0.23 |
0.73 ± 0.17 |
0.73 ± 0.24 |
0.73 ± 0.27 |
| 2 |
1.43 ± 0.25 |
1.26 ± 0.22 |
1.43 ± 0.18 |
1.08 ± 0.21 |
1.43 ± 0.23 |
0.91 ± 0.26 |
1.26 ± 0.24 |
0.91 ± 0.28 |
| 3 |
8.14 ± 0.74 |
7.13 ± 0.96 |
7.51 ± 1.03 |
6.88 ± 1.02 |
6.76 ± 0.95 |
6.76 ± 0.84 |
6.63 ± 1.07 |
6.88 ± 1.09 |
| 4 |
20.78 ±1.19 |
18.78 ± 1.38 |
19.78 ± 1.24 |
17.40 ± 1.07 |
18.28 ± 1.17 |
17.15 ± 1.09 |
17.27 ± 1.25 |
17.02 ± 1.44 |
| 6 |
38.05 ± 1.26 |
36.05 ± 1.44 |
37.55 ± 1.28 |
32.29 ± 1.36 |
34.92 ± 1.66 |
31.29 ± 1.54 |
32.04 ± 1.47 |
30.92 ± 1.69 |
| 8 |
51.32 ± 1.85 |
49.19 ± 1.65 |
49.82 ± 1.49 |
45.44 ± 1.66 |
48.57 ± 1.74 |
43.31 ± 1.95 |
44.94 ± 1.14 |
42.93 ± 1.54 |
| 12 |
64.34 ± 1.52 |
61.46 ± 1.42 |
63.59 ± 1.63 |
56.95 ± 1.85 |
60.08 ± 1.27 |
53.82 ± 1.43 |
55.45 ± 1.57 |
52.57 ± 1.62 |
| 18 |
77.98 ± 2.11 |
74.35 ± 2.09 |
76.86 ±1.96 |
68.85 ± 1.87 |
72.10 ± 2.03 |
66.47 ± 1.91 |
67.97 ± 1.88 |
64.84 ± 2.04 |
| 24 |
93.63 ± 2.66 |
86.75 ± 2.54 |
89.50 ± 1.55 |
80.24 ± 2.23 |
84.74 ± 1.45 |
77.23 ± 1.66 |
79.61 ± 1.27 |
74.35 ± 1.87 |
Table 14: In-vitro drug release from vildagliptin SR matrix tablets.

Figure 6: In vitro drug release profile of vildagliptin SR matrix
tablets.
Drug Release Kinetic Studies
The drug release data was applied into different kinetic
equations to get respective linear plots with Regression (R2)
values (Table 15). The results obtained through kinetic
treatment of dissolution data have confirmed a sustained and
controlled manner drug release from all prepared matrix
tablets. The drug release form all eight vildagliptin matrix
formulations was found to be concentration dependent
because of resulted higher R2 values for first order plots. In
case of all eight formulations, more linearity has been noticed
with Hixson-Crowell followed by Higuchi plots with slight
difference. So, the drug release from all vildagliptin matrix
formulations can be through surface erosion followed by
diffusion mechanisms.
| Formulation |
Zero order |
First order |
Higuchi |
Korsemeyer-Peppas |
Hixson-Crowell |
| R2 |
R2 |
R2 |
R2 |
n |
R2 |
| V1 |
0.9325 |
0.9721 |
0.9731 |
0.9357 |
1.7268 |
0.9884 |
| V2 |
0.9247 |
0.992 |
0.9689 |
0.9234 |
1.6678 |
0.9825 |
| V3 |
0.9264 |
0.9916 |
0.9708 |
0.9378 |
1.7344 |
0.9866 |
| V4 |
0.9269 |
0.9903 |
0.9703 |
0.9301 |
1.7649 |
0.9769 |
| V5 |
0.9236 |
0.9905 |
0.9681 |
0.931 |
1.6425 |
0.9792 |
| V6 |
0.9294 |
0.9897 |
0.9721 |
0.916 |
1.9852 |
0.9762 |
| V7 |
0.929 |
0.9904 |
0.9711 |
0.9309 |
1.7678 |
0.9777 |
| V8 |
0.923 |
0.9848 |
0.9696 |
0.9099 |
1.6442 |
0.9693 |
Table 15: Kinetic treatment for vildagliptin release data.
Randomized 23 Full Factorial Optimization and
Mathematical Modeling
The obtained in vitro drug release data (cumulative %
drug release after 24 hours) of eight formulations was
subjected to ANOVA test using DOE software to identify significant terms (with p<0.05) used in multiple
regression equation. The selected modified model used in
software for ANOVA testing was proved to be significant
(p<0.05) (Table 16).
| Source |
Sum of squares |
Degree of freedom |
Mean square |
F-value |
p-value |
|
| Model |
299.03 |
6 |
49.84 |
23592.48 |
0.005 |
Significant |
| A-Amount of HRLM |
104.47 |
1 |
104.47 |
49454.92 |
0.0029 |
| B-Amount of ZP |
43.48 |
1 |
43.48 |
20581.21 |
0.0044 |
| C-Amount of TSP |
146.12 |
1 |
146.12 |
69169 |
0.0024 |
| AC |
1.42 |
1 |
1.42 |
672.01 |
0.0245 |
| BC |
0.8646 |
1 |
0.8646 |
409.28 |
0.0314 |
| ABC |
2.68 |
1 |
2.68 |
1268.46 |
0.0179 |
| Residual |
0.0021 |
1 |
0.0021 |
|
| Cor Total |
299.04 |
7 |
|
Table 16: ANOVA for selected factorial model (Vildagliptin formulations).
The final regression equation containing only significant terms
along with their respective estimated coefficients was as
follows:
Y=83.26–3.61A–2.33B–4.27C+0.421AC+0.329 BC+0.579 ABC
The significant factors showing considerable effect on
response were A, B, C and AC, BC, ABC. The value and sign of
estimated coefficient possessed by each significant term are
deciding tools for magnitude and direction of respective
influence on response (Y). All main individual factors like A, B
and C have shown the negative effect on ‘Y’ i.e. the decreased
drug release has been resulted with an individual increase in
any polymer quantity due to enriched cross linking within a
formulation. The interaction factors like AC, BC and ABC have
exerted the positive influence on response. An increased
interaction of polymers present in a dosage form has led to
enhanced drug release which may be due to enriched
hydrophilicity and porosity. The contribution of factor C (TSP
quantity) to decrease the drug release was 48.86%. The other
main factors like A and B have contributed towards reduction
of drug release up to 34.94% and 14.54%, respectively. The
contribution of all three interaction terms towards
enhancement of response was comparatively very less (Table
17).
| Term |
Standardized effect |
% Contribution |
| A-Amount of HRLM |
-7.23 |
34.94 |
| B-Amount of ZP |
-4.66 |
14.54 |
| C-Amount of TSP |
-8.55 |
48.86 |
| AC |
0.8425 |
0.4747 |
| BC |
0.6575 |
0.2891 |
| ABC |
1.16 |
0.8961 |
Table 17: Contribution of significant terms towards response (Vildagliptin release).
The effect of factors like ‘A and C’ and ‘B and C’ on
‘cumulative vildagliptin release after 24 hours’ was visualized
through Contour and 3D surface plots which were drawn by
keeping other third polymer at low level (60 mg). All these
plots have indicated the maximum drug release (93.63%) from
a formulation composition having each polymer at its lower
level (60 mg) (Figures 7-10).

Figure 7: Contour plot showing effect of A and C on
vildagliptin release.

Figure 8: 3D Surface plot showing effect of A and C on
vildagliptin release.

Figure 9: Contour plot showing effect of B and C on
vildagliptin release.

Figure 10: 3D Surface plot showing effect o f B a nd C on
vildagliptin release.
The effect of three polymers on response (Y) was visualized
by drawing cube plot (Figure 11). This plot also pointed
the maximum drug release at a composition having all
three polymers at low level (60 mg).

Figure 11: Cube plot showing effect of A, B and C on
vildagliptin release.
All the plots drawn using modified statistical model have
suggested a best optimized formulation composition which
consisted of three polymers like HRLM, ZP and TSP, each at 60
mg quantity. This composition was same as that of developed
V1 formulation. The statistical model has suggested a
predicted cumulative drug release of 93.64% from optimized
formulation. The predicted value was found to be very close
to observed drug release response from V1 formulation
(93.63%). So, V1 formulation was confirmed as best SR matrix
tablet dosage form for vildagliptin and further used for
stability and in vivo studies.
In vitro Drug Release: Marketed Versus Optimized
Formulation
A complete vildagliptin release has been noticed from
marketed product (Vysov, manufactured by Cipla Ltd.) within
6 hrs. The pattern of drug release was immediate and not in
sustained manner from marketed formulation. But the objective of extended drug release was fulfilled by V1
formulation by releasing the drug slowly up to 24 hours
(Figure 12). The promised sustained action and prolonged
drug residence time can be expected from developed V1
formulation because of delayed release. So, it can be said that
the V1 formulation may avoid the risk of frequent dosing
which is practiced with Vysov tablets.

Figure 12: Comparative vildagliptin re lease profile of marketed
and V1 formulations.
Stability Studies
No physical and chemical degradation of best
optimized tablets (V1) has been noticed up to 180 days
(Tables 18-20). The tested tablets have retained their
important properties like drug content, swelling index and
drug release during stability study (at three different
stability conditions). It was confirmed that the tablets of
selected optimized formulation (V1) can have long term
stability at storage conditions.
| Parameter |
Initial |
After 30 days |
After 60 days |
After 90 days |
After 180 days |
| Appearance |
Appropriate |
Appropriate |
Appropriate |
Appropriate |
Appropriate |
| Weight (mg) |
402.46 ± 1.27 |
402.63 ± 0.91 |
401.49 ± 0.79 |
401.73 ± 0.83 |
402.49 ± 0.67 |
| Hardness (kg/cm2) |
4.2 ± 0.33 |
4.2 ± 0.16 |
4.1 ± 0.31 |
4.2 ± 0.11 |
4.1 ± 0.24 |
| Thickness (mm) |
3.47 ± 0.21 |
3.48 ± 0.19 |
3.48 ± 0.31 |
3.47 ± 0.22 |
3.49 ± 0.19 |
| Friability (%) |
0.69 ± 0.14 |
0.67 ± 0.14 |
0.68 ± 0.19 |
0.66 ± 0.23 |
0.68 ± 0.13 |
| Drug content (%) |
101.06 ± 1.03 |
100.76 ± 1.14 |
99.94 ± 1.77 |
99.67 ± 1.47 |
99.85 ± 1.53 |
| Swelling Index (%) |
182.36 ± 3.29 |
183.35 ± 1.66 |
185.48 ± 1.91 |
181.43 ± 1.09 |
182.67 ± 1.73 |
| CDR after 24 hrs (%) |
93.63 ± 2.66 |
94.66 ± 1.01 |
92.34 ± 1.16 |
93.49 ± 1.27 |
94.67 ± 1.81 |
Table 18: Stability data of optimized Vildagliptin formulation (V1) at room temperature.
| Parameter |
Initial |
After 30 days |
After 60 days |
After 90 days |
After 180 days |
| Appearance |
Appropriate |
Appropriate |
Appropriate |
Appropriate |
Appropriate |
| Weight (mg) |
402.46 ± 1.27 |
403.46 ± 1.06 |
401.82 ± 1.61 |
403.71 ± 1.38 |
402.46 ± 0.97 |
| Hardness (kg/cm2) |
4.2 ± 0.33 |
4.3 ± 0.27 |
4.2 ± 0.17 |
4.1 ± 0.19 |
4.2 ± 0.11 |
| Thickness (mm) |
3.47 ± 0.21 |
3.48 ± 0.07 |
3.47 ± 0.09 |
3.48 ± 0.11 |
3.48 ± 0.08 |
| Friability (%) |
0.69 ± 0.14 |
0.69 ± 0.13 |
0.68 ± 0.11 |
0.67 ± 0.14 |
0.66 ± 0.29 |
| Drug content (%) |
101.06 ± 1.03 |
100.32 ± 1.22 |
100.21 ± 1.19 |
99.93 ± 1.34 |
98.63 ± 2.08 |
| Swelling index (%) |
182.36 ± 3.29 |
183.28 ± 1.14 |
181.52 ± 1.67 |
184.91 ± 1.97 |
181.64 ± 1.27 |
| CDR after 24 hrs (%) |
93.63 ± 2.66 |
93.61 ± 1.04 |
94.82 ± 1.27 |
94.01 ± 1.03 |
95.08 ± 0.77 |
Table 19: Stability data of optimized Vildagliptin formulation (V1) at 30°C/60% RH.
| Parameter |
Initial |
After 30 days |
After 60 days |
After 90 days |
After 180 days |
| Appearance |
Appropriate |
Appropriate |
Appropriate |
Appropriate |
Appropriate |
| Weight (mg) |
402.46 ± 1.27 |
402.44 ± 1.27 |
400.64 ± 1.98 |
401.61 ± 1.67 |
400.74 ± 2.07 |
| Hardness (kg/cm2) |
4.2 ± 0.33 |
4.3 ± 0.12 |
4.2 ± 0.09 |
4.3 ± 0.11 |
4.1 ± 0.13 |
| Thickness (mm) |
3.47 ± 0.21 |
3.44 ± 0.37 |
3.46 ± 0.18 |
3.49 ± 0.16 |
3.47 ± 0.03 |
| Friability (%) |
0.69 ± 0.14 |
0.67 ± 0.14 |
0.68 ± 0.15 |
0.69 ± 0.07 |
0.68 ± 0.12 |
| Drug content (%) |
101.06 ± 1.03 |
99.16 ± 1.46 |
101.67 ± 1.41 |
100.19 ± 1.81 |
99.67 ± 1.31 |
| Swelling index (%) |
182.36 ± 3.29 |
183.46 ± 1.37 |
184.35 ± 1.17 |
182.67 ± 1.06 |
185.49 ± 1.64 |
| CDR after 24 hrs (%) |
93.63 ± 2.66 |
95.11 ± 1.94 |
94.69 ± 1.79 |
94.86 ± 1.36 |
93.91 ± 1.49 |
Table 20: Stability data of optimized vildagliptin formulation (V1) at 40°C/75% RH.
In vivo studies
The chromatogram obtained through developed HPLC
method as two peaks at different retention times
corresponding to both vildagliptin and internal standard. A
good linearity as per Beer-Lambert law has been seen with
constructed standard graph of vildagliptin in rabbit plasma.
The generated drug concentration-time profile using
constructed calibration curve is presented in Figures 13-15.

Figure 13: HPLC chromatogram of vildagliptin.

Figure 14: Standard graph of vildagliptin in Rabbit plasma
(HPLC method).

Figure 15: Plasma vildagliptin concentration-time profiles in
three rabbit groups.
All the estimated PK parameters were presented in Table 21.
The mean Cmax value in reference group was high and sharp
(693.97 ± 11.81 ng/ml) compared to other two groups. The
plasma drug concentration has reached to its maximum
after 1 hour of dosing in reference group. The rapid
drug absorption from reference solution has been confirmed
by this result. The Tmax values for marketed product and
V1 formulation were 2 hours and 4 hours, respectively. The
rate of drug absorption from marketed tablet was also
rapid and next to reference solution. But, the rate of
vildagliptin absorption from optimized formulation (V1) was
delayed due to extended drug release from dosage form.
More mean values of AUC (6799.28 ± 54.63 ng.hr/ml) and
MRT (18.6 ± 1.19 hrs) were resulted from third group
which received V1 formulation compared to other two
groups. The rabbits of first and second groups have
shown mean AUC values as 3867.77 ± 42.16 ng.hr/ml
and 5831.46 ± 37.19 ng.hr/ml, respectively. MRT values
resulted from reference solution and Vysov table were
comparatively less which may be due to rapid drug
absorption and elimination. An increased oral
bioavailability of drug from optimized formulation has been
noticed and it may be due to enhanced residence time. The
most convincing and promising benefits have been noticed
with developed V1 formulation compared to conventional
marketed product [18].
| PK parameter |
Reference |
Marketed product (Vysov) |
Optimized formulation (V1) |
| Cmax (ng/ml) |
693.97 ± 11.81 |
512.56 ± 23.41 |
427.37 ± 13.44* |
| Tmax (hrs) |
1.0 ± 0 |
1.5 ± 0 |
4.0 ± 0 |
| AUC0-t (ng.hr/ml) |
3867.77 ± 42.16 |
5831.46 ± 37.19 |
6799.28 ± 54.63* |
| MRT (hrs) |
3.4 ± 0.69 |
4.8 ± 0.44 |
18.6 ± 1.19* |
| All values expressed as Mean ± SD; *Significant with p<0.05. |
Table 21: Pharmacokinetic parameters of vildagliptin in rabbits.
Conclusion
In vitro drug release from all developed sustained release
matrix tablets was influenced by quantity of each polymer
accommodated inside the dosage form. The extents of some
deciding properties like swelling index, matrix forming ability
and release retarding capacity were found to be more with
TSP polymer followed by HRLM and ZP, respectively. The
identified best optimized formulation was V1 (for vildagliptin)
with a formulation composition consisting of all three
polymers like HRLM, ZP and TSP, each at their lower level (60
mg). The results of stability studies and in vivo tests using
animal models were satisfactory for these best formulations.
The estimated pharmacokinetic parameters of drug for each
best formulation were in support of obtained in vitro drug
release profile indicating a good ‘In Vitro In-Vivo Correlation’ (IVIVC). Further research is required and
recommended for future in vivo studies using human models
to estimate all required pharmacokinetic parameters and to
compare the same with obtained results in animal models.
References
- Nihar R, Pani L, Nath K (2014) Development of controlled release tablet by optimizing HPMC: Consideration of theoretical release and RSM, Carbohydrate polymers. Int J Pharm Sci. 104:238-245.
[Crossref] [Google Scholar] [PubMed]
- Jyosna D, Jeevana J (2020) Novel tamarind seed gum-alginate based multi particulates for sustained release of dalfampridine using response surface methodology. Int J Biol Macromol. 144:725-741.
[Crossref] [Google Scholar] [PubMed]
- Madhu SR, Dilshad Q, Monjurul H, Girija MPJ, Biswaranjan M, et al. (2020) Synthesis and characterization of novel tamarind gum and rice bran oil based emulgels for the ocular delivery of antibiotics. Int J Biol Macromol. 164:1608-1620.
[Crossref] [Google Scholar] [PubMed]
- Gourang N, Amit K, Nandi N, Sarif K, Souvik P, et al. (2018) Tamarind seed gum hydrolyzed poly methacryl amide-g-gellan beads for extended release of diclofenac sodium using 32 full factorial design. Int J Biol Macromo. 114:214-225.
[Crossref] [Google Scholar] [PubMed]
- Sanjay D, Bankim CN, Jayanta ND, Saquib HMd, Amit KN (2019) Tamarind gum in drug delivery applications. Nat Polys Drug Deliv Bio Appli. 19:285-306.
[Crossref] [Google Scholar]
- Sachiko F, Hideki Y, Shuichi Y, Suyoshi MT, Hidemi M, et al. Design and evaluation of an extended release matrix tablet formulation the combination of hypromellose acetate succinate and hydroxyl propyl cellulose. Asian J Pharm Sci. 12(17):149-156.
[Crossref] [Google Scholar] [PubMed]
- Alena K, Vaclav L, Katerina M, Jitka M, Martin B (2016) Formulation and dissolution kinetics study of hydrophilic matrix tablets with tramadol hydrochloride and different co-processed dry binders. Eur J Pharm Sci. 95:36-45.
[Crossref] [Google Scholar] [PubMed]
- Jishnu V, Gilhotra RM, Prabhakaran R (2011) Formulation and evaluation of cephalexin extended release matrix tablets using 32 factorial design. J Young Pharm. 3(4):259-266.
[Crossref] [Google Scholar] [PubMed]
- Nagasamy V, Meyyanathan SN, Shanmugam R, Zielinska A, Campos JR, et al. (2020) Development, in vitro release and in vivo bioavailability of sustained release nateglinide tablets. J Drug Deliv Sci Technol. 55:101-105.
[Google Scholar]
- Manodeep C, Mohammed GA, Ananya B (2017) Potential pharmacodynamic and pharmacokinetic interaction of pomegranate juice and nateglinide against diabetis induced complications in rats. Synergy. 5:1-6.
[Crossref] [Google Scholar]
- Mona F, Arafa SA. El-Gizawy M, Osman GA, Maghraby ME (2017) Co-crystallization for enhanced dissolution rate of nateglinide: In vitro and in vivo evaluation. J Drug Deliv Sci Technol. 38:9-17.
[Crossref] [Google Scholar]
- Pushkar R, Sharma SA. Lewis, Design and in vitro/in vivo evaluation of extended release matrix tablets of Nateglinide. J Young Pharm. 5(4):167-172.
[Crossref] [Google Scholar] [PubMed]
- Chisato M, Hidetoshi S, Akira O, Akira Y (2009) Design of nateglinide controlled release tablet containing erosion matrix tablet and multiple administration study in normal beagle dogs. Chem Pharm Bull. 57(9):907-913.
[Crossref] [Google Scholar] [PubMed]
- Pushkar S, Shaila L (2013) Design and in vitro/in vivo evaluation of extended release matrix tablets of Nateglinide. J Young Pharm. 5(4):167-172.
[Crossref] [Google Scholar] [PubMed].
- Sathyaseelan V (2013) Formulation development and in vitro evaluation of sustained release matrix tablets of Nateglinide by using natural polymers. J Young Pharm. 132:186-193.
[Google Scholar]
- Sudheer BB, Pamula R, Swamy PV, Mohan VV, Basava RD, et al. (2001) Dose calculation, design and development of nateglinide matrix tablets using quality by design approach and its pharmacokinetic evaluation in animal model. J Pharm Investig. 45(6):515-528.
[Google Scholar]
- Ameena K, Dilip C, Saraswathi R, Krishnan PN, Sankar C, et al. (2010) Isolation of the mucilages from Hibiscus Rosa Sinensis linn. and Okra (Abelmoschus esculentus linn.) and studies of the binding effects of the mucilages. Asian Pac J Trop Med. 3(7):539-543.
[Crossref] [Google Scholar]
- Ashwini R, Madgulkar MR, Bhalekar VJ, Kolhe YD (2009) Formulation and optimization of sustained release tablets of Venlafaxine resinates using response surface methodology. Indian J Pharm Sci. 71(4):387-394.
[Crossref] [Google Scholar] [PubMed]
Citation: Deepika B, Sujatha K (2023) Design and Evaluation of Vildagliptin Matrix Tablets by Response Surface Methodology. Am
J Drug Deliv Ther. 10:11.
Copyright: © 2023 Deepika B, et al. This is an open-access article distributed under the terms of the Creative Commons
Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author
and source are credited.

