Keywords
Antioxidants; Glucosephosphate Dehydrogenase; Polyphenols
INTRODUCTION
Glucose 6 phosphate dehydrogenase (G6PD) catalyzes the first step in the pentose phosphate pathway (conversion of glucose 6-phosphate to 6-phosphogluconate) with the generation of NADPH required for the maintenance of reduced glutathione and for bile acid synthesis [1].
The NADPH that is provided by this enzyme is the main source of reducing equivalents for the lipogenic pathway thereby making this enzyme a crucial lipogenic enzyme [2]. G6PD also plays an important role in preventing oxidative damage to human cells by participating in the generation of antioxidants. The NADPH generated by this pathway is used as an electron donor for multiple intracellular reactions such as the regeneration of reduced glutathione (GSH), an important intracellular antioxidant, thereby offering protection to cells against oxidative stress [3].
Umesh et al. [4] reported a decrease in the hepatic glucose- 6-phosphate dehydrogenase activity of streptozotocin diabetic rats. Similarly, Debidas et al. [5] reported that a reduction in the G6PD activity in diabetic rats contributed to a reduction in metabolism via the gluconate oxidative pathway.
In our previous study [6], we described for the first time, the anti-diabetic potentials of Livingstone potato in streptozotocin induced diabetic rat models. However, there is no experimental evidence presently available in literature with regard to its effect on the G6PD activity of diabetic rats.
Since supplementation of therapeutics with plant based antioxidants/polyphenols could have chemoprotective role in the management of diabetes mellitus [7] and in a bid to further understand the mechanism of anti-diabetic action of Livingstone potato, this study was set out to investigate the phytochemical composition, antioxidant capacity and effect of Livingstone potato on the hepatic G6PD activity of streptozotocin diabetic rat models.
MATERIALS AND METHODS
Plant Materials
The Plectranthus esculenta varieties were obtained at harvest from National Root Crops Research Institute, Umudike, Nigeria. They were identified by NRCRI, Umudike that has Livingstone potato as a National Mandate as well as by a Taxonomist in Michael Okpara University of Agriculture, Umudike, Nigeria and deposited in their herbarium for authentication.
Chemicals
Streptozotocin, β-Nicotinamide Adenine Dinucleotide Phosphate-Sodium Salt, Triethanolamine Hydrochloride, Glucose-6-phosphate monosodium salt, magnesium chloride salt, 2,2-diphenyl-1-picryl hydrazyl (DPPH) radical and Quercetin used were products of Sigma and Aldrich Chemical Company, United Kingdom. Other chemicals used were purchased from Hoslab, Umuahia, Abia State, Nigeria and were of analytical grade.
Processing of the Plant Materials
The samples were properly washed, chopped and oven dried at 60oC for 48 hours to constant weight. The dry samples were then processed to flour and incorporated into the standard rat feeds at 19.55% incorporation.
ANIMAL EXPERIMENTS
Selection of Animals
Thirty male albino rats of the wistar strain (140-208g) obtained from the animal house of the Department of Biochemistry, University of Nigeria, Nsukka, Enugu State, Nigeria were used for the study. The animals were kept in metabolic cages in the animal house of the Department of Biochemistry, Michael Okpara University of Agriculture, Umudike, Nigeria. They were acclimatized for two weeks to their diets prior to the commencement of the experiment and were maintained under a constant 12-h light and dark cycle and a room temperature of 27-30°C. The National Institutes of Health Principles of Laboratory Animal Care [8] were observed.
Induction of Diabetes
Freshly prepared solution of streptozotocin (0.1g dissolved in 5 ml of freshly prepared sodium citrate buffer 0.1 M, pH 4.5) was injected intraperitoneally to the rats at a dosage of 65mg/kg body weight at fasting state. Blood was collected from the tail vein and the blood glucose concentration was analyzed prior to the commencement of the dietary feeding using a blood glucose meter (Double G glucometer, USA). The STZ-treated rats with fasting blood glucose levels > 200 mg/dl after seven days of induction of STZ and with evidence of glycosuria were considered to be diabetic and used for the study.
Experimental Procedure
The experimental rats with stable diabetic condition were then divided into 2 subgroups (groups 2 and 3) comprising of six animals per group while the non-diabetic group formed the first group as follows:
Group 1. Normal rats fed standard rat pellets (Non-diabetic control)
Group 2. Diabetic control rats fed standard pellets
Group 3. Diabetic rats fed Livingstone potato incorporated feeds.
BIOCHEMICAL ANALYSIS
Their diets and water were both administered ad libitum for 28days after which the rats were stunned by blow, sacrificed and their liver was removed, washed with icecold physiological saline immediately and stored at -20oC until analyzed. Ten percent homogenate (w/v) of the liver was prepared in 150 mM KCl using a homogenizer at 4°C and centrifuged for 15 min at 4°C (9) and the supernatant was analyzed for glucose-6-phosphate dehydrogenase activity. The specific activity of NADP linked glucose- 6-phosphate dehydrogenase activity (EC 1.1.1.49) was determined using the method of Noltmann et al. [9, 10].
DPPH Radical Scavenging Assay
The method of Blois [11] was used with modifications. A measured amount (5 g) of each sample was dissolved in 100ml of methanol to give a stock concentration of 50mg/ ml and left for 3 to 4 hrs. The mixture was filtered with Whatmann No 1 filter paper through vacuum pump.
The resulting solution was diluted with methanol to final concentrations of 50, 100, 150, 200 and 250μg/ml respectively. Finally, 0.1ml of 0.3mM DPPH (in methanol) was added to each of the reaction mixtures and the whole setup was well shaken and left in the dark for 30mins before the absorbance was read spectrophotometrically at 517nm against the DPPH control that contained 1 ml of methanol only in place of the extract. The percentage scavenging activity was calculated as:
[(Absorbance of control - Absorbance of sample)/ Absorbance of control]*100.
Quercetin was used as the standard for this assay.
Reducing Power Assay
The method of Hsu et al. [12] was used with modification. A measured amount (2 g) of each sample was dissolved in 40 ml of methanol to give a stock concentration of 50mg/ ml and left for 3 to 4 hrs. It was filtered with Whatmann No 1 filter paper through vacuum pump, made up to 50 ml with methanol to give a concentration of 40 mg/ml and diluted to suitable concentrations: 5, 10, 15, 20 and 25 mg/ml for reducing power assay. One ml of each extract + 0.5 ml of phosphate buffer (0.2 M, pH 6.6) and 2.5 ml of potassium hexacyanoferrate solution (1% in water) were placed in a test tube and reacted for 20 min at 50oC. The tubes were cooled with crushed ice and an aliquot of 0.5 ml trichloroacetic acid (10% in water) was added. After centrifugation at 3000 g for 10 min, an aliquot (1 ml) of the supernatant was mixed with an equal volume of distilled water + 0.1 ml of ferric chloride (0.1% in water) and the set up was allowed to react for 10 min before the absorbance was measured at 700nm using a UV spectrophotometer. Increased absorbance of the reaction mixture indicates increased reducing power. Quercetin was used as the standard.
PHYTOCHEMICAL ANALYSIS
Determination of Alkaloids
The gravimetric method of Harborne [13] was used in the determination of total alkaloid content of the test feed. Five gram of each sample was dispersed into 50ml of 10% acetic acid solution in ethanol. The mixture was well shaken and allowed to stand for 4hours before filtering. It was evaporated to one quarter of its original volume and drop wise concentration of ammonium hydroxide was added to precipitate the alkaloids. The precipitate was filtered off with a pre-weighed filter paper and washed with 1% ammonium hydroxide solution. The precipitate was oven dried for 30 minutes at 60oC and re-weighed. The alkaloid content of the samples was determined by difference using the equation:
Percentage alkaloid = (W2 – W1)/W*100
Where W = Weight of sample; W1 = Weight of empty filter paper; W2 = Weight of paper + precipitate.
Determination of Flavonoid Content
Ten grams of each sample was extracted with 100 ml of 80% aqueous methanol at room temperature for 2 to 4 hours. The mixture was filtered through Whatmann No 1 filter paper. The filtrate was later transferred into a Petri dish, evaporated to dryness over a water bath (70oC) and weighed to constant weight [14].
Determination of Cyanide Content
The alkaline picrate method [15] was employed for the determination of cyanide contents of the test feed. A portion (5 g) of each sample was grinded into paste and dissolved in 50 ml distilled water in a corked conical flask. The extraction was allowed to stay for 12 hrs. The sample was filtered and the filtrate was used for cyanide determination. To 1 ml of the sample filtrate in a corked test tube, 4 ml of alkaline picrate (prepared by dissolving 1g of picric acid in 5g of Na2CO3 and made up to 100ml with H2O) was added and incubated in a water bath for 5 minutes for colour development (reddish brown) and the absorbance was read at 490 nm after stabilizing the spectrophotometer with the absorbance of the blank (which contained 1 ml of distilled water and 4 ml of alkaline pirate solution). The cyanide content was extrapolated from a cyanide standard curve and the cyanide content was calculated using the formula:
Cyanide (mg/kg) = Absorbance* Gradient Factor* Dilution Factor / Weight of the sample
Determination of Tannins
The percentage composition of tannin in the test feed was determined using the method of AOAC [16]. The tannin content of the samples was calculated from a tannic acid standard curve and results were expressed as milligrams of tannic acid equivalence (TAE) per 100 g of dried sample.
Determination of Saponins
Saponin determination was done using the method of AOAC [16]. Saponin extraction was done using two different solvents. The first solvent, acetone, was used to extract crude lipid from the samples while the second solvent (methanol) was used for the extraction of the saponin proper. Two gram of the sample was folded into a thimble and put in a sox let extractor and a reflux condenser fitted on top. Extraction was done with acetone in a 250cm3 capacity round bottomed flask for 3 hours, after which the apparatus was dismantled and another 150cm3 capacity round bottomed flask containing 100cm3 of methanol was fitted to the extractor and extraction was carried on for another 3 hours. The weight of the flask was taken before and after the second extraction in order to make the change in weight. At the end of the second extraction, the methanol was recovered by distillation and the flask was oven-dried to remove any remaining solvent in the flask. The flask was then allowed to cool and the weight of the flask taken. The saponin content of the sample was calculated as:
% Saponin = Weight of saponin / Weight of sample*100
STATISTICS
Data was subjected to analysis using the Statistical Package for Social Sciences (SPSS), version 15.0. Results were presented as the means ± standard deviations of duplicate or triplicate experiments. One way analysis of variance (ANOVA) was used for comparison of the means. Differences between means were considered to be significant at P < 0.05 using the Duncan Multiple Range Test.
RESULTS
The hepatic G6PD activities of the non-diabetic control, diabetic control and diabetic rats administered Livingstone potato incorporated feeds were 2.80±0.37, 1.69±0.69 and 2.21±0.42 respectively. There were significant increases (P<0.05) in the G6PD activities of the diabetic rats fed the test feeds compared with the diabetic control rats while the G6PD activities of the diabetic control rats were significantly lower (P<0.05) than that of the non-diabetic rats (Figure 1).
Figure 1. Glucose-6-phosphate dehydrogenase activity of rats. Values
are the means ±SD of three determinations. abcMeans with different
superscripts for each parameter are significantly different (P<0.05): Unit
definition: G6PD- μmol of NADP+ reduced/min/g protein.
Chemical analysis of the test feed revealed that it contained 0.24±0.02% saponin, 0.28±0.02 mg/kg HCN, 0.44±0.01% alkaloid and 0.43±0.03% tannin (Figure 2).
Figure 2. Phytochemical composition of livingstone potato incorporated
feed. Values are the means ± SD of triplicate experiments.
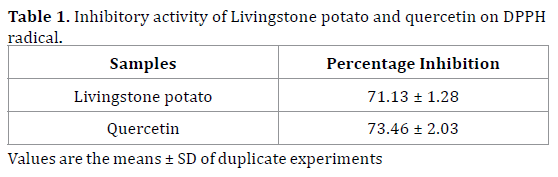
The percentage scavenging activity of the Livingstone potato incorporated feed on DPPH free radical was 71.13±1.28 which was close to that of standard quercetin (73.46 ± 2.03) (Table 1).
The reducing power of the Livingstone potato incorporated feed on the average was 0.797±0.025 which was lower than that of standard quercetin (1.190 ± 0.328) (Figure 3).
Figure 3. Reducing power of test feed and standard quercetin.
DISCUSSION
The decrease in the hepatic glucose-6-phosphate dehydrogenase activity of the diabetic untreated rats as observed in this study, could lead to decreased generation of NADPH needed for the regeneration of glutathione and other reducing agents needed for the maintenance of tissue integrity and oxidative balance, thereby leading to the induction of oxidative stress [17, 18].
One of the pathogenic mechanisms that explain the development of diabetic complications is oxidative stress resulting from increased generation of free radicals and impaired antioxidant defense system in diabetic conditions. Inhibition of these oxidative processes could prevent the onset and development of long-term diabetic complications [19, 20].
Thus, the increased activity of the hepatic G6PD activity of the diabetic rats given Livingstone potato incorporated feed is an indication of improvement in glucose utilization by the pentose phosphate pathway and also confirms our previous reports [6] that the major mechanism of anti-diabetic action of Livingstone potato is through its antioxidant scavenging mechanism.
Alkaloids and tannins are polyphenolic compounds with antioxidant properties. There are indications that apart from the scavenging of free radicals, polyphenols could also act by direct interactions with important cellular receptors or key signaling pathways leading to the modification of the redox status of cells which results in the triggering of a series of redox-dependent reactions [21]. In addition, tannins exert their action on carbohydrate metabolism through the inhibition of α-glucosidase and α-amylase, the key enzymes responsible for the digestion of dietary carbohydrates to glucose [19].Thus the considerable amount of these polyphenolic compounds in the Livingstone potato incorporated feed could be one explanation for the anti-diabetic potentials of Livingstone potato as previously reported [6].
Saponins are known to possess both beneficial (cholesterol lowering) and deleterious (cytotoxic permeabilization of the intestine) properties [22]. The saponin content of the Livingstone potato incorporated feed as observed in this study could be one explanation for the antihypercholesterolemic and anti-hyperlipidaemic action of Livingstone potato as previously reported [6].
Cyanogenic glucosides are compounds that yield glucose, hydrogen cyanide and aldehyde or ketone upon hydrolysis with an acid or enzyme. The safe level of cyanide in foods as given by WHO/FAO is 10ppm (10mg/kg) [23]. According to Bolhius [24], 50-100mgHCN equivalent kg-1 cyanide was set as moderately toxic and above 100mg HCN equivalent kg-1 as dangerously toxic or lethal. Values obtained for cyanide in the Livingstone potato incorporated feed as observed in this study, were below toxic levels.
The DPPH assay is a widely accepted method for the determination of the antioxidant activities of various food systems. The method is widely used due to the relatively short time required for the analysis. As observed in this study, the methanolic extract of the Livingstone potato incorporated feed contained strong antioxidant activity which was close to standard quercetin.
The DPPH assay is limited to neutral and higher pH applications and also suffers from colour interference [25] and this informed the assay of the reducing power of the test feeds.
Reducing power assay is another widely accepted method that is employed in the assay of the antioxidant activities of various plants and it employs the reduction of Fe3+ to Fe2+ through electron transfer [26, 27]. The methanolic extract of the Livingstone potato incorporated feed was also observed to possess a strong reducing power, indicative of strong antioxidant activity though lower than the reducing power of standard quercetin and this strong antioxidant activity is attributed to the polyphenolic constituents of Livingstone potato.
Since several studies have implicated oxidative stress in the etiology of diabetes and its complications and inhibition of these oxidative processes by plant based antioxidants could prevent the onset and development of long-term diabetic complications [20], it is plausible to attribute the modulatory action of Livingstone potato on the glucose- 6-phosphate dehydrogenase activity of diabetic rats to its antioxidant/polyphenolic constituents.
CONCLUSION
The study reveals the ameliorating action of Livingstone potato on hepatic G6PD activity of diabetic rat models which is attributed to its antioxidant/polyphenolic constituents.
Conflicts of Interest
The authors have no potential conflicts of interest.
Acknowledgement
The assistance rendered by the technical staff of the Food Chemistry Laboratory, Michael Okpara University of Agriculture, Umudike, Nigeria is acknowledged.
References
- McAnuff MA, Omoruyi FO, Morrison EY, Asemota HN. Changes in Some Liver Enzymes in Streptozotocin-induced Diabetic Rats fed Sapogenin Extract from Bitter Yam (Dioscoreapolygonoides) or Commercial Diosgenin. West Indian Med J. 2005; 54: 97. [PMID: 15999877]
- Robert KM, Darry KG, Peter AM. Harper’s Biochemistry, 23rd Edition. Appleton and Lange, USA, 1993; 150-151. [PMID:6039601]
- Jablonska-Skwiecinska E, Lewandowska I, Plochocka D, Topczewski J, Zimowski JG, Klopocka J, Burzynska B. Several mutations including two novel mutations of the glucose-6-phosphate deficient subjects with chronic nonspherocytic hemolytic anemia and favism. Hum Mutat. 1999; 14: 477-484. [PMID:10571945]
- Yadav UC, Moorthy K, Baquer NZ. Effects of Sodium-Orthovanadate and TrigonellaFoenum-Graecum Seeds On Hepatic And renal lipogenic enzymes and lipid profile during alloxan diabetes. J Biosci. 2004; 29: 12. [PMID:15286407]
- DebidasG,Tushar KK, Chatterjee K, Debasis D. Anti-diabetic and anti-oxidative effects of aqueous extract of seed of psoraleaCorylifolia (somraji) and seed of trigonellafoenum-graecum in separate and composite manner in streptozotocin-induced diabetic male albino rat. Int J Pharm Res Develop. 2009; 7: 976-984.
- Eleazu, C.O., Eleazu, K.C., Chukwuma, S.C., Ironkwe, C., Iroaganachi, M.A. and Emelike, C.U. Effect of Livingstone potato (Plectranthusesculentus N.E.Br) on diabetes and its complications in Streptozotocin induced diabetes in rats. DMJ DiabMetabol J. 2014.
- Gomathi D, Ravikumar G, Kalaiselvi M, Devaki K, Uma C. Efficacy of Evolvulusalsinoides (L.) L. on insulin and antioxidants activity in pancreas of streptozotocin induced diabetic rats. J Diabetes MetabDisord. 2013; 12:39. [PMID:23834750]
- National Research Council (NRC). Guide for the care and use of laboratory animals. Publication 8523, National Institute of Health, Bethesda, Md, USA, 1985.
- Singh SN, Vats P, Suri S, Shyam R, Kumria MM, Ranganathan S, Sridharan K. Effect of an antidiabetic extract of Catharanthusroseus on enzymic activities in streptozotocin induced diabetic rats. J Ethnopharmacol. 2001; 76: 269-277. [PMID:11448549]
- Noltmann EA, Gubler CJ, Kuby SA. Glucose 6-phosphate dehydrogenase (Zwischenferment). I. Isolation of the crystalline enzyme from yeast. J BiolChem 1961; 236:1225-1230. [PMID:13729473]
- Blois MS. Antioxidant determination by use of stable free radicals. Nature. 1985; 29:1199-1200.
- Hsu CL, Chen W, Weng YM, Tseng CY. Chemical composition, physical properties, and antioxidant activities of yam flours as affected by different drying methods. Food Chem. 2003; 83: 85-92.
- Harbone JB. Comparative Biochemistry of the Flavonoids. New York Academic Press 1973; 221-222.
- Boham AB, Kocipai AC. Flavonoid and condensed tannins from leaves of Hawaiian vaccininumvaticulum and vicalycinium. Pacific Sci. 1994; 48: 458-463.
- Onwuka GI. Food Analysis and Instrumentation. Theory and Practice. Napthali Prints 2005; 140-146.
- AOAC. Official Methods of Analysis, 15th edn. Association of Official Analytical Chemists, Arlington, VA. 1990.
- Gupta D, Raju J, Prakash J, Baquer NZ. Change in the lipid profile, lipogenic and related enzymes in the livers of experimental diabetic rats: effect of insulin and vanadate. Diabetes Res ClinPract. 1999; 46: 1-7. [PMID:10580609]
- Raju J, Gupta D, Rao AR, Yadava PK, Baquer NZ. Trigonellafoenum-graecum (fenugreek) seed powder improves glucose homeostasis in alloxan diabetic rat tissues by reversing the altered glycolytic, gluconeogenic and lipogenic enzymes. Mol Cell Biochem. 2001; 229: 22445-22451. [PMID:11693199]
- Bahadoran Z, Mirmiran P, Azizi F. Dietary polyphenols as potential nutraceuticals in management of diabetes: a review. J Diabetes MetabDisord. 2013; 12: 43. [PMID:23938049]
- Shin SH. Oxidative stress and diabetic vascular complications. Recent Advances In Pathogenesis and Management of Diabetes Mellitus. 1st edn. Elsvier Science Company, Singapore. 1998; 633: 3-8.
- Scalbert A, Johnson IT, Saltmarsh M. Polyphenols: antioxidants and beyond. Am J ClinNutr 2005; 81: 215S-217S. [PMID:15640483]
- Price KR, Johnson IT, Fenwic GR. The chemical and biological significance of saponins in food and feeding stuff. Crit Rev Food SciNutr. 1987; 26: 27-135. [PMID:3308321]
- FAO/IFAD. The World Cassava Economy: Facts, Trends and Outlooks. Food and Agriculture Organization of the United Nations and International Fund for Agricultural Development, Rome. 2000.
- Bolhius GG. The toxicity of cassava roots. Neth J Agric. 1954; 2: 176-185.
- Akiyama H, Toida T, et al . Determination of cyanide and thiocyanate in SugihirataKe Mushroom using HPLC method with fluorimetric detection. J Health Sci. 2006; 52: 73-77.
- Choong CT, Van-Den T, Roger FM, Roger LT, Kenneth VP, Craig Y. Antioxidant activities, phenolic and ß-carotene contents of sweet potato genotypes with varying flesh colours. Food Chem. 2007; 103: 829-838.
- Parameswari G, Suriyavathana M. Evaluation of total phenolic content and free radical scavenging activity of Boerhaviaerecta. J Acute Med 2013; 2: 1-7.




