Keywords
Heavy metals; Reactive oxygen species; Oxidative stress; Antioxidants; Renal damage
Introduction
Heavy metals are metals with relatively high densities, atomic weights or atomic numbers, and include Hg, Pb, As, cadmium (Cd), nickel (Ni), chromium (Cr), Uranium (U) and thallium (Tl) [1,2].
Anthropogenic activities cause global contamination and pollution, the resultant health of which is heightened by the bioaccumulation of metal ions in the environment and in humans [3,4]. Most heavy metals at high concentrations exhibit harmful effects on the central nervous system (CNS), energy metabolism, ion transporters, cardiovascular system, respiratory system, reproductive system and vital organs such as lungs, liver, brain and kidney [5,6]. The combination of heavy metals with other xenobiotics such as pesticides exerts synergistic toxic effects on hematology and the immune system [7]. However, the degree of toxicity depends on factors such as route of exposure, number of exposures, time of exposure, type of metal ion, duration of exposure, age, health status, nutrition and genetic makeup of the individual [8]. Exposure to heavy metals such as Pb, and decreases in the levels of essential metals such as selenium (Se) and Zn cause oxidative stress. This results in a shift in the redox status of the cell, thereby causing damage to biomolecules such as lipids, nucleic acid and proteins, as well as organs such as liver, kidney and CNS [9,10]. Aerobic organisms are the main targets of oxidative modifications that result in uncontrolled generation of ROS such as superoxide anion (O2-), hydroxyl radical (HO-), hydrogen peroxide (H2O2), peroxyl radical (ROO-) and reactive aldehydes (ROCH+) [11,12]. An enzyme complex of nitric oxide synthases (NOSs) consisting of neuronal NOS type 1, inducible NOS (iNOS) type 2, endothelial NOS (eNOS) type 3 and mitochondrial NOS (mtNOS) catalyzes the synthesis of various nitrogen products [13]. For instance, iNOS catalyzes the synthesis of nitric oxide (NO.) from L-arginine, which in turn reacts with ROS to form several RNS [14]. The RNS are involved in oxidation and nitrosation reactions, and they comprise NO., nitrogen dioxide (NO2), dinitrogen trioxide (N2O3), dinitrogen tetroxide (N2O4) and peroxynitrite (ONOO.) [14,15]. Both ROS and RNS exert different effects on mediators of cell signaling cascades, such as those involved in necrosis and neurodegenerative disorders [16,17]. Apart from exogenous sources, ROS and RNS are also produced endogenously via partial reduction of molecular oxygen (O2) in the respiratory chain within the mitochondria [18]. Metabolic alteration caused by inflammatory processes or interaction of molecules mediating ROS synthesis can significantly elevate the levels of ROS and RNS [12].
Studies have shown that low levels of ROS and RNS support physiological processes such as cell proliferation, host defense, signal transduction and gene expression [11]. Eukaryotic cells have several antioxidant defense mechanisms such as enzymes and antioxidant biomolecules that maintain cellular balance between the generation of ROS and RNS, and their elimination [16]. Antioxidants are low molecular weight molecules that protect intracellular components against the deleterious effects of ROS/RNS. Indeed, all aerobic organisms have developed highly efficient antioxidant strategies throughout evolution [19]. However, when the cellular production of ROS/RNS exceeds the intrinsic antioxidant capacity, oxidative stress occurs, which causes damage to intracellular and extracellular biomolecules, resulting in tissue degeneration [20]. In addition, ROS/RNS are generated by exposure to ultraviolet light, ionizing radiation or exposure to heavy metal ions [16-20]. The combined effect of excessive production of ROS/RNS and reduced antioxidant capacity leads to oxidative stress, loss of cell function and cell death induced by apoptosis [19]. The oxidant/antioxidant cellular imbalance leads to a vicious circle, since oxidative stress reciprocally aggravates ROS/RNS production [20]. Oxidative stress is also responsible for the activation of various transcription factors, leading to differential expression of genes involved in inflammatory pathways [12]. Mitochondrial DNA (mtDNA) is particularly susceptible to oxidative damage in contrast to its nuclear equivalent, due to its proximity to the internal mitochondrial membrane [11]. Another factor that influences mtDNA susceptibility to oxidative damage is the lack of histones and other associated proteins, the absence of introns, and high rate of transcription in its coding region [12]. Oxidative stress-induced DNA damage affects the coding region of mtDNA and influences oxidative phosphorylation, thereby altering the respiratory chain and unregulated production of ROS [11]. This phenomenon in mtDNA is one of the causes of cellular oxidative stress. One of the cellular adaptations employed by organisms in the presence of oxidative stress is the modification of the ratio of the activity of glutathione peroxidase to the level of glutathione disulfide (GPx/GSSG) [20,21]. This ratio is regulated by the major antioxidant enzymes: superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) [19]. Glutathione peroxidase (GPx) catalyzes the removal of hydroxyperoxides via the modulation of enzyme-reduced glutathione (GSH) [22]. The expressions of GPx and CAT are modulated by hydrogen peroxide concentration and exogenous factors such as exposure to heavy metals [23].
Chronic kidney disease is progressive, and it is characterized by the loss of functional nephrons, and/or structural, molecular and functional changes in nephrons. As the disease develops, there is reduction in glomerular filtration and failure of the remaining functional nephrons to efficiently eliminate metabolic and toxic wastes from the body. Heavy metals such as Pb, Hg and U have harmful effects on the kidney, particularly on the proximal tubules, which suggests that the nephron plays an important role in the active transport of heavy metals [24,25]. Oxidative stress can occur in the glomeruli, interstitial tubule and renal vasculature. It has been implicated in many processes such as the expansión of glomerular mesangium, nephropathy events, increased glomerular filtration, excretion of urinary albumin, proteinuria, glomerulosclerosis and tubular interstitial fibrosis [26]. In renal biopsies, the glomerulus exhibits extensive sclerosis, chronic glomerular ischemia, tubular interstitial fibrosis and mild vascular lesions [27]. The present study comprehensively reviewed the effects of Hg, Pb, As and Zn on renal oxidative status.
Methods
Literature survey was done using key words such as heavy metals, oxidative stress and kidney damage. The search engines used were free scientific publications such as PubMed database, FreeFullPDF.com and Google Scholar. In addition, reference lists from relevant original articles and documents were manually reviewed.
Results
Mercury and oxidative stress
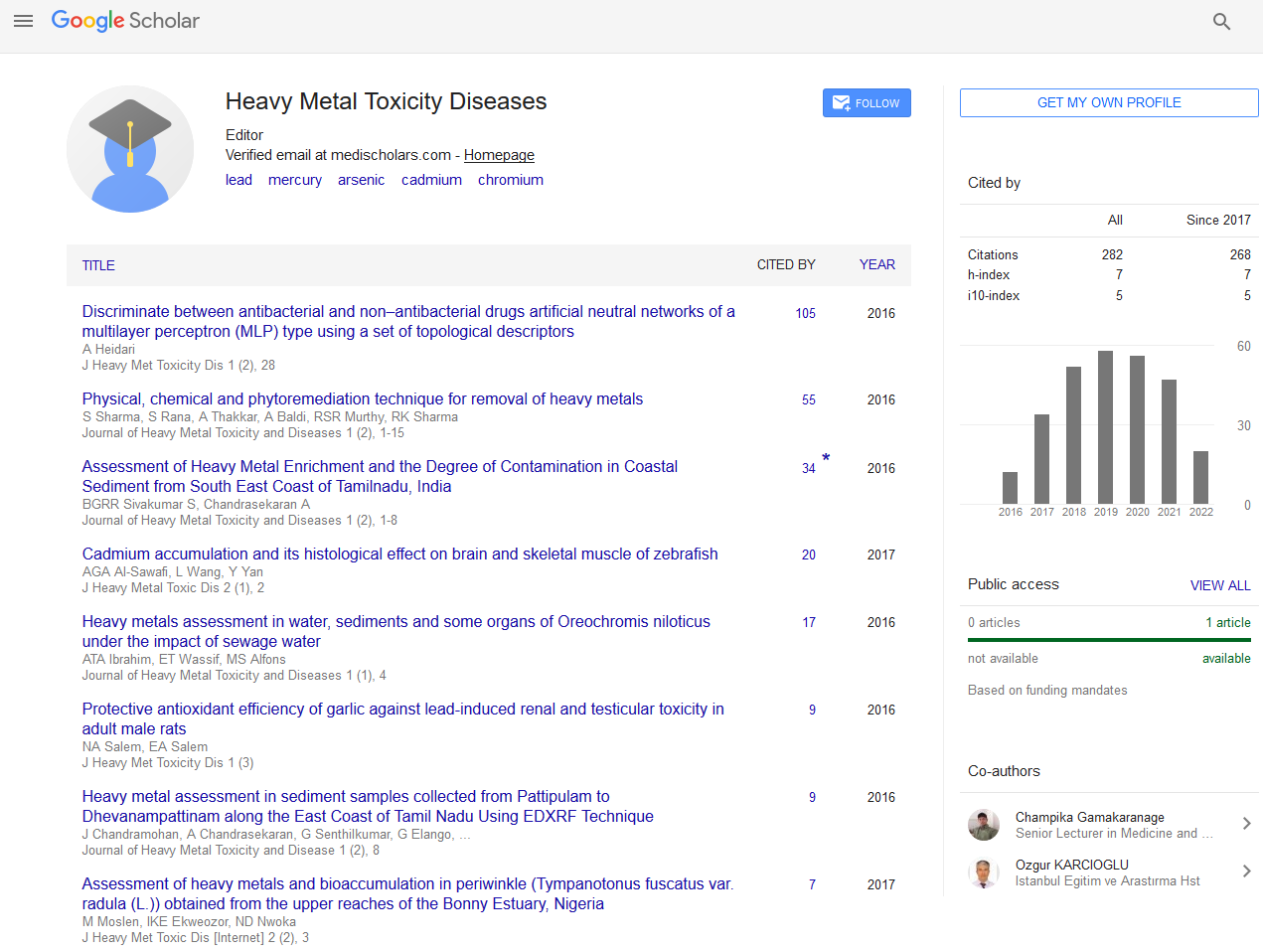
Mercury is a toxic metal which exist in different forms: elemental (metallic), inorganic and organic. Elemental Hg is liquid at room temperature [1]. Inorganic Hg exist as ions (Hg+ or Hg2+) which binds to chloride, sulfur or oxygen to form mercuric salts [28]. The organic forms of mercury are phenylmercury (C6H5Hg), dimethylmercury ((CH3)2Hg) and methylmercury (CH3Hg +). Methylmercury is the most common in the environment and it is formed when Hg+ or Hg2+ is methylated by the activities of microorganisms present in soil and water [29]. Human exposure to mercury is linked to anthropogenic activities [1]. Mercury is one of the earliest elements found to be non-essential for biological processes; it is considered toxic due to its accumulation in organisms [30]. Mercury exerts its effect at the neurological, renal, respiratory, immunological, dermatological, reproductive and developmental levels [28]. Mercury toxicity varies depending on the chemical form involved, and its route of exposure [28]. Mercury induces oxidative stress which causes membrane damage, enzymatic damage and oxidation of biomolecules [3]. It has been reported that exposure to mercury decreases the catalytic activity of GPx in rats, and promotes the synthesis of H2O2 and lipid peroxidation (LPO) products in renal and mitochondrial membranes [31-33]. Thus, mercury stimulates the production of malondialdehyde (MDA), 4-hydroxyalkenes (4-HOA) and advanced protein oxidation products such as dityrosine, which increases the inflammatory response (Figure 1).
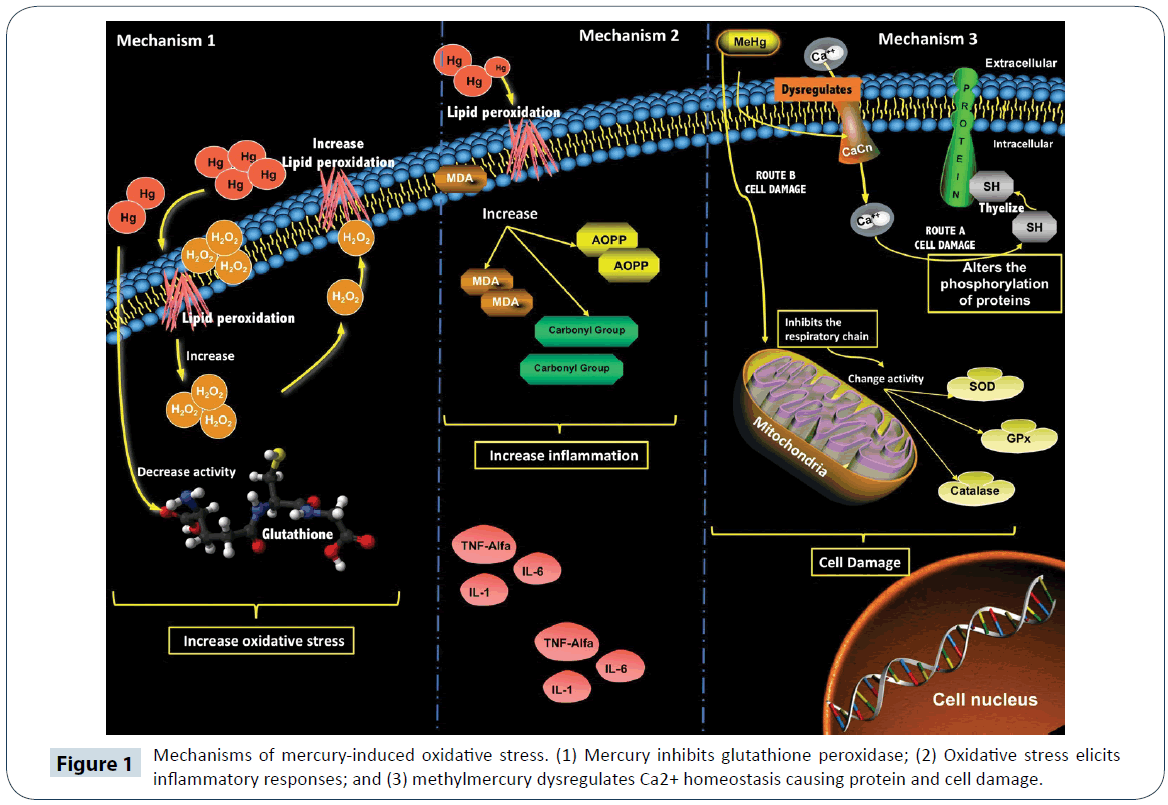
Figure 1: Mechanisms of mercury-induced oxidative stress. (1) Mercury inhibits glutathione peroxidase; (2) Oxidative stress elicits inflammatory responses; and (3) methylmercury dysregulates Ca2+ homeostasis causing protein and cell damage.
Methylmercury (CH3Hg+) is neurotoxic: it damages microtubules and mitochondria; it modulation of the accumulation of biomolecules that are neurotoxic at high concentrations, such as serotonin, glutamate (Glu) and aspartate (Asp), and it impairs intracellular Ca2+ homeostasis. Moreover, methylmercury alters protein phosphorylation, stimulates thiol group (-SH) binding which damages cellular structure, interrupts the cell cycle, and binds to proteins rich in cysteine or methionine [32-34]. In neurons, (CH3Hg+) inhibits the uptake of astrocytic Glu and then stimulates its exit from the cytosol, leading to an increase in Glu level in the extracellular fluid [35]. Glutamate targets N-methyl- D-aspartate receptor in the brain, and stimulates increases in Na+ and Ca2+ levels in neuronal cells. Calcium ion (Ca2+) at high concentrations, acts as a second messenger that alters protein phosphorylation [36-38]. On the other hand, CH3Hg+ directly interrupts mitochondrial activity via uncontrolled release of Ca2+ from the mitochondria, and inhibition of the phosphorylation and function of mitochondrial enzymes [39]. In one study, CH3Hg+ inhibited the mitochondrial electron transport chain in vitro [39]. In another study, it was found that CH3Hg+ had an inhibitory effect on GPx activity in mouse CNS, which resulted in an increase in LPO products and a decrease in Glu uptake in cerebral cortex [40]. It has also been reported that NO- production after microglial activation causes decreases in cellular GSH levels. It is a known fact that CH3Hg+ has high affinity for GSH, which results in significant reduction in its level [41,42]. In the mitochondria, CH3Hg+ increases the action potential of the inner membrane which regulates HO-, O2- and H2O2, and negatively regulates the defense enzymes SOD, CAT, GPx, as well as GSH (Figure 1) [18].
Mercury and human disease
The neurotoxic effect of Hg is progressive and destructive, especially in the central and peripheral nervous systems of children. Vapors contaminated with Hg can cause chemical pneumonitis, necrotizing bronchitis, asthma and respiratory problems such as cough and dyspnea [43]. The early symptom of Hg intoxication in children is pruritic rash which is similar to dermatitis, but it can also cause Hunter-Russell syndrome and Minamata disease [4]. Prolonged exposure to Hg damages the brain and causes timidity, tremors, memory problems, irritability, and changes in hearing and vision. Brief exposure to metallic Hg vapors at high temperatures causes lung damage, vomiting, diarrhea, nausea, skin rashes, increased cardiac output and elevated blood pressure [43]. Symptoms of organic Hg poisoning include depression, memory problems, tremors, fatigue, and headache and hair loss [2]. The United States Environmental Protection Agency (EPA) has declared mercury chloride (HgCl2) and CH3Hg+ as carcinogens [44]. Mercury inhibits the enzyme catechol orthomethyltransferase via inactivation of its coenzyme S-adenosyl methionine (SAM), thereby producing high concentrations of catecholamines with symptoms almost indistinguishable from those of pheochromocytoma [34].
In vitro studies involving exposure to CH3Hg+ have revealed dysfunction in the biochemical processes that trigger Alzheimer's disease and axonal degeneration [45,46]. This may be due to the fact that CH3Hg+ has the highest body distribution in humans, when compared with other Hg derivatives. In humans, CH3Hg+ intoxication leads to reduction of visual field, bronchitis, pneumonitis, tremors, salivation and intense gingivitis [45]. It has also been shown that high intake of Hg through non-fat products is associated with an increased risk of heart failure, arrhythmias, myocardial infarction, coronary heart disease and cardiovascular disease [47]. Animal studies have shown that increased exposure to various Hg species affects and inhibits immune function [48]. High Hg levels affects the relative abundance of immune cells and the production of cytokine signals, while low Hg levels affects cytokine signals without modifying the immune cell count [49].
Mercury and renal failure
The various forms of Hg are nephrotoxic. However, exposure to Hg2+ conjugated with Pb produces more severe nephropathy, and the proximal tubules appear to be the most sensitive, while other segments are only affected at high doses [29]. Analysis using electron microscopy have shown that 12 h after exposure to HgCl2, the cells of the renal tubules had ruptured plasma membranes, loss of microvilli, decreased contact with the basement membrane and destruction of the cell morphology [34]. In addition, chronic exposure of rats to non-nephrotoxic dose of HgCl2 led to tubular, interstitial and glomerular lesions [50]. Mercury salts affect mainly the gastrointestinal tract (GIT) and kidneys, and cause acute tubular necrosis, immunological glomerulonephritis or nephrotic syndrome after prolonged exposure. This is because Hg ions preferentially accumulate within epithelial cells of renal tubules. Therefore, high Hg levels lead to kidney injury [43]. It has been reported that chronic exposure to inorganic Hg, elemental mercury vapors and ingestion of Hg2+ salts result in nephrotic syndrome characterized by albuminuria, proteinuria or acute tubular necrosis [44]. On the other hand, exposure to metallic Hg is not usually of great concern since it is malabsorbed via the intestine. However, in one study, it was reported that a 67-year old patient on ingestion of metallic Hg developed severe pneumonitis and acute renal failure. Although the amount ingested was unknown, the serum concentration was extremely high. However, normal renal function was recovered after thirteen episodes of hemodialysis [24]. However, studies with Wistar rats have shown that administration of of CH3Hg+ at a dose of 0.04 mg/kg body weight for 35 days was able to produce deposits of this metal and induction of oxidative stress at the salivary gland level [51]. Similarly, in rats chronically exposed to of CH3Hg+, fibrotic changes were observed in the glomeruli, and deposition of immunoglobulin G (IgG), immunoglobulin M (IgM) and complement component C3 were detected along the glomerular basement membrane [52]. These results suggest that chronic exposure to Hg2+ or of CH3Hg+ may lead to the development of membranous glomerulonephritis. Glomerular alterations such as fibrosis and glomerulonephritis often lead to reductions in glomerular filtration [53,54]. Recently, an epidemic of renal failure in Central America (Mesoamerican nephropathy) was reported. This has been described mainly in farmers who are clinically asymptomatic, with normal or slightly high blood pressure, and progressive reduction in glomerular filtration, proteinuria in the non-nephrotic range (<3 g/24 h), hyperuricemia and hypocalcemia [55]. Tissue histology revealed interstitial tubule disruption or glomerular ischemia with secondary glomerulosclerosis [55]. Several risk factors have been proposed for this disease, including occupational exposure to heavy metals such as Hg and pesticides [56].
Lead and oxidative stress
Lead (Pb), the fifth most used metal worldwide, is toxic to humans, and its toxicity is related to the induction of oxidative stress [57,58]. The physiological damage caused by Pb depends on the route of exposure, age of the subject, health status; number of exposure, time of exposure and genetic makeup of the individual [59]. Absorption of Pb in the inorganic state is facilitated by the respiratory chain and GIT [60]. Lead acetate, (CH3CH2)4Pb, the most organic Pb, is an anti-knock compound usually added to automobile fuel. It is absorbed mainly via the respiratory tract. Organic lead is more soluble than inorganic Pb, due to the high lipid-solubility characteristic of its components which facilitates its distribution within organs and tissues [60]. Once it gets to the bloodstream, 95 to 99 % of Pb binds to hemoglobin, and spreads throughout the body. The half-life of Pb is approximately 30 days, prior to its excretion mainly in urine [23]. The toxicity of Pb depends largely on its dose. For example, in healthy adults weighing 70 kg, plasma level ranging from 10 to 30 mg/dL inhibits delta-aminolevulinic acid synthase (δ-ALAS), δ-aminolevulinic acid dehydratase (δ-ALAD), coproporphyrinogen decarboxylase, ferrochelatase and pyrimidine 5-nucleotidase (Figure 1). The inhibition of these enzymes leads to oxidative stress [23,57]. The enzyme δ-ALAS is important for the production of aminolevulinic acid, which is bioprocessed to protoporphyrin IX in mitochondria, and then chelated with iron to form heme [61]. On the other hand, δ-ALAD catalyzes the condensation of aminolevulinic acid to form tetrapyrroles, chlorophyll and vitamin B12, and it is highly sensitive to Pb exposure [62]. Oxidative stress is also a consequence of the inhibition of enzymes such as δ-ALAS and δ-ALAD. It has been reported that δ-ALAD autooxidation, which generate O2- anions and 4, 5 dioxovaleric acid, an alkylating agent of guanine residues within the DNA [23]. The autooxidation form of ALAD acts as an electron donor, capable of transferring an electron to oxygen transported in oxyhemoglobin to form methemoglobin, ALAD-radical and H2O2 [63]. Subsequently, the interaction of O2- with H2O2 generates HO-, a powerful oxidant of lipids, proteins and DNA (Figure 2) [63]. In addition, Pb has high affinity for -SH group and metal cofactors, which leads to reduction in the activity of antioxidant enzymes [23]. In animal models, exposure to lead acetate led to inhibition of ALAS in blood, activation of plasma CAT, elevation of MDA and GSSG, reduction in the level of plasma GSH, and an increase in oxygen free radicals (Figure 1) [64]. These results are consistent with those of Ercal et al., who reported significant reductions in the levels of GSH and increases in GSSG and MDA levels, relative to the control group [65]. It is likely that exposure to Pb led to inhibition of ALAS, which increased the accumulation of ALAD capable of auto-oxidizing ROO- and HO-, while promoting glutathione oxidation [64,66]. Doses of Pb lower than 80 mg/dl stimulate the production of H2O2, thereby alter the expression of GPx. At higher doses, Pb displaces selenocysteine group from the active site of glutathione, and decreases the binding of substrate to the -SH groups, thereby weakening enzyme-substrate interaction, resulting in reduced activity of GPx [23]. In a previous study, the activity of GPx decreased up to 77% in rats exposed to Pb, when compared with the control group [22]. Redox status (GSH/GSSG) is an index of oxidative stress in mammals [66,67]. With regard to the enzymatic response of SOD to Pb exposure, a positive regulation has been found at increased concentrations of O2- [23]. In addition, MDA products are enhanced with Pb exposure, which inactivates SOD genes [23]. On the other hand, in one study, an increase in the activity of SOD was observed in individuals exposed to Pb, possibly as a result of adaptation to ROS [66]. Similarly, in another study involving workers exposed to Pb in Sao Paulo, there were increased activities of SOD and GPx, when compared with non-exposed persons [67]. There was a 148% increase in SOD activity in G. polyedra cells exposed to Pb for 2 h [68]. In Wistar rats exposed to Pb, the renal Cu/Zn- SOD activity decreased significantly by 61%, relative to the group without exposure [22]. In rats exposed to Pb, the activity of renal CAT decreased by 34%, when compared with control group [22], an indication that CAT may modulate GPx. However, the activities of GPx and CAT, and concentration of MDA were significantly increased with increase in plasma concentration of Pb [69].
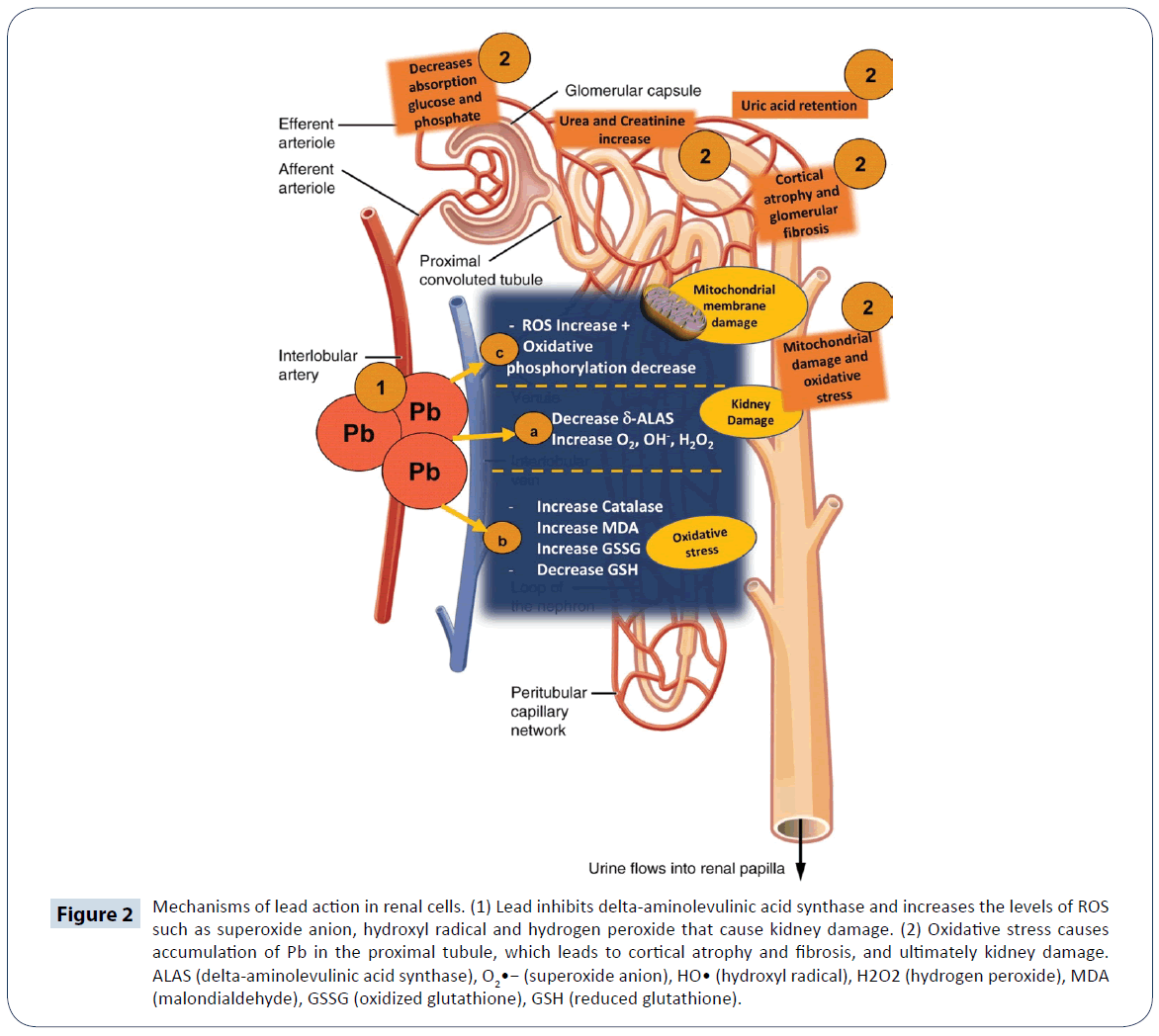
Figure 2: Mechanisms of lead action in renal cells. (1) Lead inhibits delta-aminolevulinic acid synthase and increases the levels of ROS such as superoxide anion, hydroxyl radical and hydrogen peroxide that cause kidney damage. (2) Oxidative stress causes accumulation of Pb in the proximal tubule, which leads to cortical atrophy and fibrosis, and ultimately kidney damage. ALAS (delta-aminolevulinic acid synthase), O2•− (superoxide anion), HO• (hydroxyl radical), H2O2 (hydrogen peroxide), MDA (malondialdehyde), GSSG (oxidized glutathione), GSH (reduced glutathione).
Lead and renal failure
Exposure to Pb is associated with kidney disease and high blood pressure [66]. Lead affects mainly the proximal convoluted tubules where it interferes with mitochondrial function, a decreases the reabsorption of glucose, amino acids and phosphate [57]. In a previous study, cortical atrophy and glomerular fibrosis were detected after exposure to Pb. Other effects of Pb on the kidney are increased blood pressure and renal failure, because of its affinity for transport along with the Na+-Ca2+-EDTA complex in the bloodstream [60]. The concentration of Pb in exposed miners is positively correlated with symptoms of nephropathy [58,59]. In adolescents exposed to (CH3CH2)4Pb in Turkey, there was positive correlation between blood levels (5 g/dL) and urinary excretion of N-acetyl-beta-D-glucosaminidase, which is an early marker of kidney injury [64]. There is no exact data on the toxic dose at renal level. However, blood concentrations of 5 to 10 mg/dL may cause tubular interstitial nephritis [63]. It has been reported that concentrations of 2.3 to 72.5 mg/dL to cause glomerular damage [22]. It has been reported that levels higher than 80 mg/ dL cause toxic effects in renal tubules due to the retention of uric acid (Figure 2) [60]. Lead-induced kidney damage is associated with increased oxidative stress and mitochondrial dysfunction, ineffective oxidative phosphorylation, interference with Ca2+- dependent reactions, mitochondrial membrane damage, and cell necrosis (Figure 2) [60,63,65,69]. Increased oxidative stress reduces the activity of guanylate cyclase, which depletes cyclic guanidine monophosphate (cGMP) and increases Ca2+ and NOlevels. This causes vascular resistance which severely affects renal blood pressure [60,63]. Increased Ca2+ levels are linked to the activation of apoptosis and acceleration of renal damage in rats [22]. In this sense, renal tissue damage may be related to the formation of Schiff bases between structural proteins and free radicals generated by ALAD [66,70]. In addition, Pb accelerates the production of LPO in the presence of Fe2+ [66], and the resultant MDA alters membrane enzyme activity and transport, modifies signaling processes, changes the permeability of mitochondrial inner membrane, enhances autoxidation of membrane lipids, and decreases SOD activity [67,70]. Elevated MDA has been linked to increased renal nitrotyrosine level [21]. These results are in agreement with results of previous studies, where it was reported that MDA levels and GPx activity are significantly higher in adolescents exposed to Pb, relative to the control group. It was reported in one study that exposure of Wistar rats to Pb produced a 117% increase in ROS in the kidney, when compared with the control group [22].
Arsenic and oxidative stress
Arsenic (As) is a metalloid which exist in four oxidation states: AsO43-, AsO2-, elemental arsenic (As0), and arsine (AsH3) [71]. The EPA classifies arsenic as a class A carcinogen [72]. It causes carcinogenesis, cytotoxicity and genotoxicity in humans under conditions of uncontrolled exposure [71]. The main routes of exposure are environmental contamination and labor contamination [73]. The AsO43- form is predominant in the air, while AsO43- and AsO2- forms are predominant in water, soil and food [74]. The degree of its toxicity depends on factors such as dose, susceptibility of the individual, age and chemical status. The toxicity of its different chemical forms follows the order: AsO43- 5AsO3)2H7AsO2)<(AsO2) [4,71]. The absorption of as depends on the route of exposure: 80 to 90% of AsO43- and AsO2- are absorbed through the GIT, and cutaneous absorption is very slow for any of its chemical forms [74,75]. Within 24 h of absorption, inorganic As binds to hemoglobin in liver, kidney, heart, lungs, CNS, spleen and GIT [75]. It is excreted mainly in urine as C2H7AsO2 3 to 5 days after exposure. It is also excreted through tissues rich in keratin [75]. The trivalent state of arsenic (As3+) is the main form that is transported to the interior of cells and this occurs via simple diffusion through aquaporins. However, its pentavalent form (As5+) requires active transport, and once inside the cell, it is reduced to trivalent arsenic [71,74]. At the cellular level, derivatives of As influence the activity of enzymes responsible for the synthesis/ degradation of heme and activation of heme oxygenase [74]. In the liver, As undergoes biomethylation reactions catalyzed by arsenite methyltransferase, with SAM serving as methyl group donor [74]. This reaction involves a series of GSH-mediated reduction-oxidation steps [76]. Arsenic is capable of binding to the -SH groups of glutathione, which modifies the proportion of GSH to GSSG, during which process H2O2 is formed [75-77]. The binding of AsO43- and AsO2- to lipoate leads to concomitant inhibition of the Krebs cycle and interference with oxidative phosphorylation. Arsenic inhibits the absorption of glucose in the cells, thereby inducing gluconeogenesis and the oxidation of fatty acids [77]. It forms ADP-arsenate complex which uncouples oxidation from phosphorylation and decreases the generation of adenosine triphosphate (ATP) (Figure 3) [76]. Therefore, this process induces apoptosis in high energy-dependent tissues [75]. Mitochondrion is the primary site of O2 radical formation during oxidative phosphorylation. Arsenic modifies the GSH/ GSSG, thereby increasing the sensitivity of cells to ROS damage [77]. The results of several studies showed that GSH level is significantly reduced after exposure to As [77-79]. For example, in one of such studies, it was found that the concentration of GSH, and activities of glucose-6-phosphate dehydrogenase (G6PDH) and GPx were significantly reduced in liver of male Wistar rats, after six months of exposure to As. The GSH acts as an electron donor in the reduction of As, with AsO2- having high affinity for GSH [78]. Therefore, arsenic-induced oxidative damage is accompanied by reduction in the level of tissue GSH [73]. Arsenic also promotes the generation of high amount of H2O2 through the Fenton and Haber-Weiss reaction [74,79]. A positive correlation has been shown to exist between ROS and renal cytotoxicity [79]. Increased level of ROS alters the activities of some antioxidant enzymes such as SOD, CAT, GPx and heme oxygenase-1, while increasing LPO, and altering the regulatory mechanisms of cell proliferation and apoptosis (Figure 3) [75]. In a previous study, other than the generation of ROS, the presence of RNS was reported to be responsible for the oxidation of lipids, proteins and DNA [73]. However, the results of some studies did not support this claim, possibly due to the down-regulation of iNOS expression [72]. Thus, the underlying mechanism by which RNS induces lipid peroxidation is still not fully elucidated. In some recent studies, it has been suggested that As may not be able to inhibit the expression of iNOS gene in muscle cells of rats exposed to As [80]. In one study involving low levels of As (<5 μM), there were no significant changes in the intracellular concentration of nitrite [77-80]. Some reports suggest that arsine (AsH3) generates intermediate species and releases H2O2 through a spontaneous and exergonic chemical reaction [77]. The signaling cascade of caspases responsible for inducing mitochondrial apoptosis is activated by ROS. The cytotoxicity produced by ROS is due to the activation of c-Jun N-terminal kinases, which promote cell proliferation, differentiation and apoptosis [14]. The release of tumor necrosis factor α (TNF α) is also stimulated by ROS via the activity of c-Jun N-terminal kinases [81].
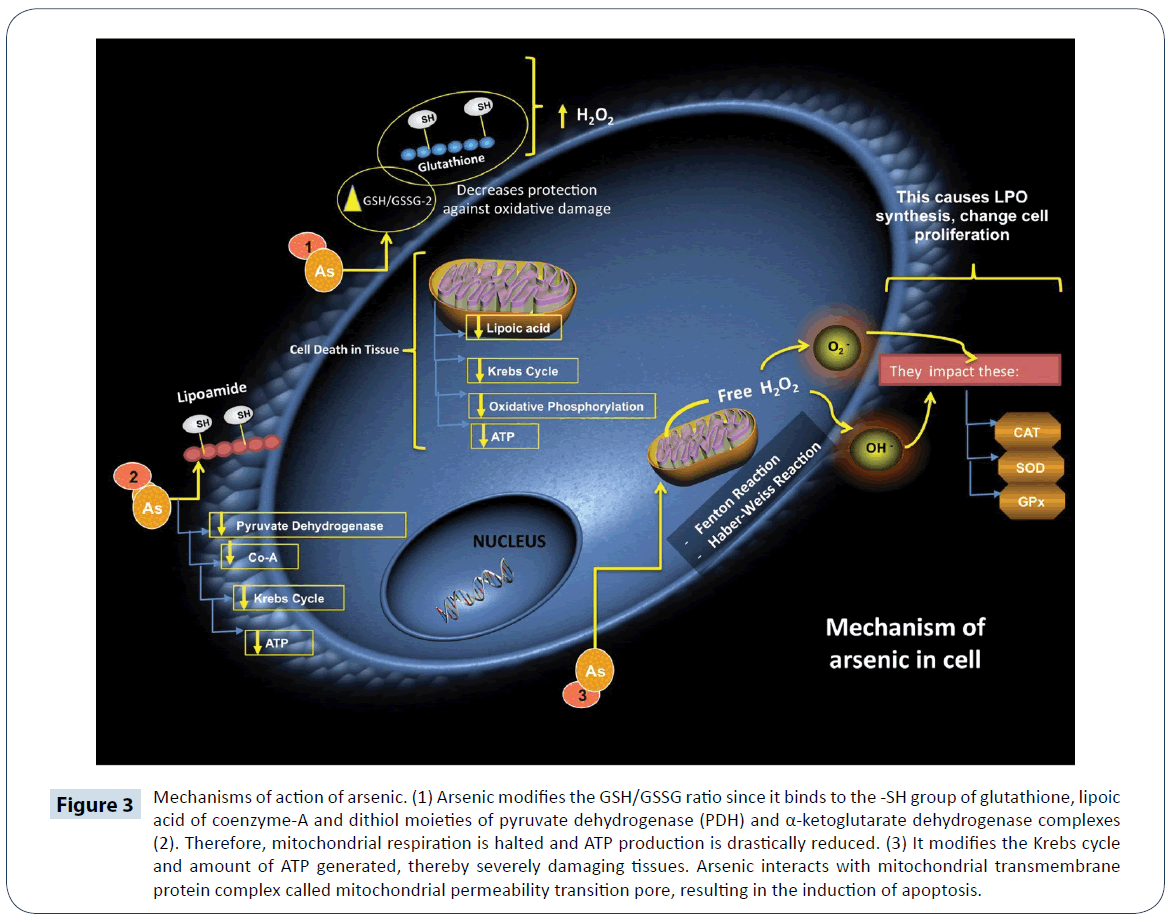
Figure 3: Mechanisms of action of arsenic. (1) Arsenic modifies the GSH/GSSG ratio since it binds to the -SH group of glutathione, lipoic acid of coenzyme-A and dithiol moieties of pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase complexes (2). Therefore, mitochondrial respiration is halted and ATP production is drastically reduced. (3) It modifies the Krebs cycle and amount of ATP generated, thereby severely damaging tissues. Arsenic interacts with mitochondrial transmembrane protein complex called mitochondrial permeability transition pore, resulting in the induction of apoptosis.
Arsenic and renal failure
The kidney is one of the organs most sensitive to As toxicity: there is a strong association between environmental exposure to As and chronic kidney disease [77,82]. Studies have shown that exposure to high doses of As produces histological changes in kidney tubules of Wistar rats, an indication that the urinary system is sensitive to damage induced by C2H7AsO2 [83]. Arsenic promotes increase in volume and pH of the urine; decreases electrolyte levels, increases Ca2+ excretion and increases in kidney weight. Moreover, it promotes degeneration of epithelial cells of the proximal convoluted tubule and tubular cylinders, enhances focal mineralization, damages podocytes of the Bowman's capsule, increases serum levels of urea and creatinine, and brings about histological changes in kidney tubules (Figure 4) [77,84]. Renal tissue has high metabolic energy requirements and depends on aerobic metabolism for ATP production via oxidative phosphorylation. Thus, the utilization of O2 in the electron transport chain (ETC) in the mitochondria is vital for renal cell function [85]. Oxidative stress and nitrosative stress reduce the enzymatic activities of complexes III, IV and V of the respiratory chain, thereby reducing the level of ATP. This causes mitochondrial dysfunction in renal cells and in turn generates a self-perpetuating vicious circle of oxidative stress. The loss of mitochondrial membrane potential releases cytochrome C through the oxidation of cardiolipin, which activates caspases and the apoptotic pathways in renal cells [84,86]. Apoptosis occurs due to the formation of pores in mitochondrial membrane, which decreases the membrane potential by releasing pro-apoptotic proteins, and causes an imbalance in cellular homeostasis [86]. Polyunsaturated fatty acids present in biological membranes are attacked by ROS resulting in an imbalance in the fluidity and integrity of membrane phospholipids [87]. Specifically, ROS attack the brush border membrane that covers epithelial cells of renal proximal tubule, and induce histopathological damage. It has been suggested that enzymes anchored within the brush border membrane are highly sensitive to decreases in activities caused by membrane LPO, since this can uncouple them from the membrane [88]. In addition, ROS significantly upregulate the expression of mitogen-activated protein kinase (MAPK) pathways that regulate transcription factor 2, and activate ETS-1pointed domain, thereby increasing renal toxicity [89]. Arsenic induces apoptosis and genotoxicity in humans by inhibiting DNA repair in renal tissue, which causes chromosomal aberrations, micronuclei formation and epigenetic modifications [88,90].
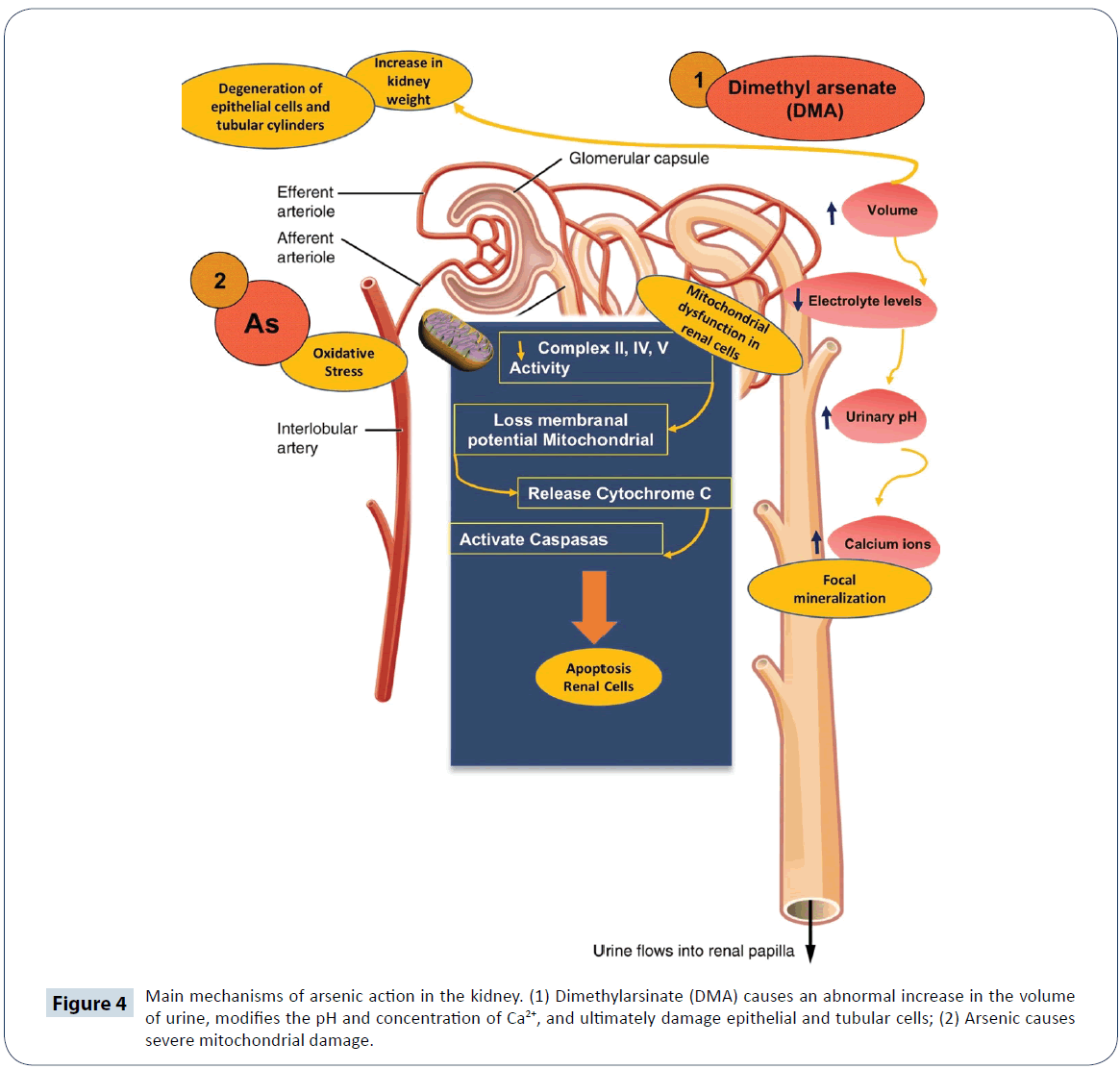
Figure 4: Main mechanisms of arsenic action in the kidney. (1) Dimethylarsinate (DMA) causes an abnormal increase in the volume of urine, modifies the pH and concentration of Ca2+, and ultimately damage epithelial and tubular cells; (2) Arsenic causes severe mitochondrial damage.
Zinc deficiency and oxidative stress
Zinc (Zn) is a trace element found in plants and animals. In mammals, Zn is a component of more than 70 different enzymes involved in the metabolism of proteins, carbohydrates and lipids [91]. Studies have shown that Zn deficiency leads to growth retardation and hypogonadism. However, its metabolic effects are not well-understood, and its diagnosis is complicated [92]. Symptoms of Zn deficiency include loss of appetite, dermatitis, delayed wound healing, deterioration of reproductive health and reduction in immune function. Severe deficiency of Zn is rare, and the most common cause is a genetic predisposition [93]. Zinc deficiency has been associated with increased oxidative damage to lipids, proteins and DNA [94]. Animal studies have shown that a prolonged deficiency of Zn leads to oxidative stress. Zinc deficiency combined with ROS production leads to lipid peroxidation in lung tissue, formation of conjugated dienes and MDA in microsomes, lipoprotein oxidation and galactosamine-induced hepatitis in rats (Figure 5) [80]. It has been reported that Zn deficiency induces a conformational change in SOD similar to mutation, thereby leading to oxidative stress in the endoplasmic reticulum, inhibition of the synthesis of some proteins and induction of the zip-14 Zn transporter [95]. Zinc is an inhibitor of N-methyl-Daspartate (NMDA) receptor which participates in Ca2+ transport to the cytosol. Therefore, Zn deficiency leads to activation of NMDA receptor and an increase in the intracellular concentration of Ca2+, which activates leukocytes and macrophages. This favors the release of pro-inflammatory cytokines, free radicals, activation of NADPH oxidase and iNOS, and concomitant production of ROS and RNS which cause oxidative stress [96,97]. Oxidative stress caused by Zn deficiency induces changes in mammary gland, which promote the infiltration of macrophages, thereby creating a microenvironment that increases the risk of breast cancer [98].
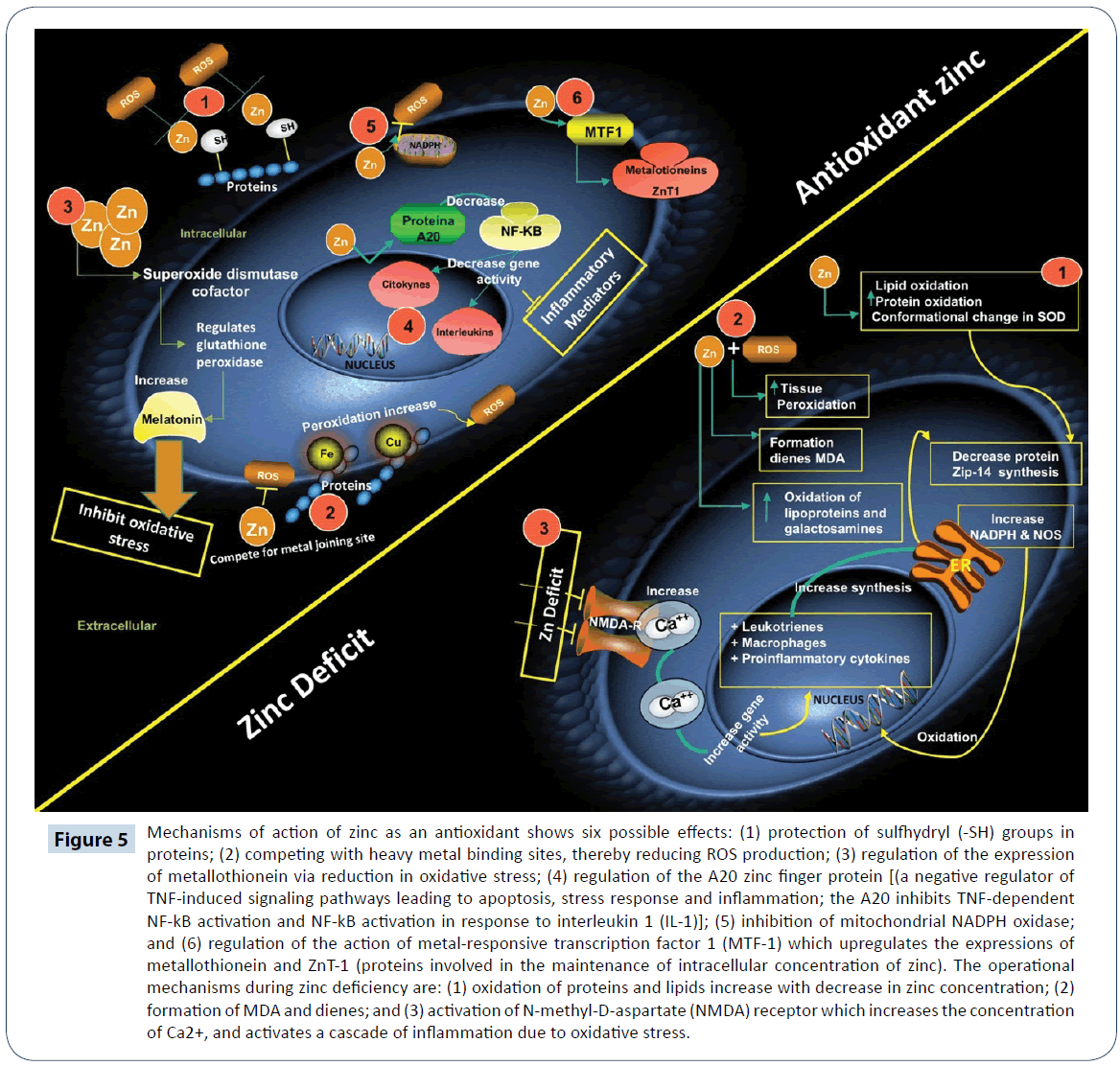
Figure 5: Mechanisms of action of zinc as an antioxidant shows six possible effects: (1) protection of sulfhydryl (-SH) groups in proteins; (2) competing with heavy metal binding sites, thereby reducing ROS production; (3) regulation of the expression of metallothionein via reduction in oxidative stress; (4) regulation of the A20 zinc finger protein [(a negative regulator of TNF-induced signaling pathways leading to apoptosis, stress response and inflammation; the A20 inhibits TNF-dependent NF-kB activation and NF-kB activation in response to interleukin 1 (IL-1)]; (5) inhibition of mitochondrial NADPH oxidase; and (6) regulation of the action of metal-responsive transcription factor 1 (MTF-1) which upregulates the expressions of metallothionein and ZnT-1 (proteins involved in the maintenance of intracellular concentration of zinc). The operational mechanisms during zinc deficiency are: (1) oxidation of proteins and lipids increase with decrease in zinc concentration; (2) formation of MDA and dienes; and (3) activation of N-methyl-D-aspartate (NMDA) receptor which increases the concentration of Ca2+, and activates a cascade of inflammation due to oxidative stress.
Oxidative stress and chronic exposure to alcohol downregulate the expressions of Zn transport proteins which alter not only Zn homeostasis, but also the function of cellular organelles (endoplasmic reticulum and mitochondria) in which the proteins are found, while Zn supplementation promotes the expression of Zn transporters: Zip-8, Zip-13 and Zn-T14 [99-101]. It has been reported that individuals with oxidative stress-induced diseases have altered functional state of Zn which makes it difficult to control such diseases. For example, an obese person with Zn deficiency experiences oxidative stress triggered by high concentration of MDA.
Zinc as an antioxidant
The function of Zn as an antioxidant may involve one of two mechanisms: (i) protection of protein -SH groups against free radicals attack; and (ii) decrease in production of free radicals via antagonism of the transition state of active metals [101]. As an antioxidant, zinc reduces the formation of free radicals in two main ways: it acts as an inhibitor of NADPH oxidase; and it reduces the production of inflammatory cytokines via regulation of A20 protein, which inhibits nuclear transcription factor kappa B (NF-κB). It has been observed that exposure of cells in vitro to high concentration of Zn is accompanied by increased expression of A20, and inhibition of the expressions of NF-κB, pro-inflammatory cytokines and interleukin, thus making Zn an anti-inflammatory agent [102,103]. Some studies have highlighted its role in the regulation of GPx activity and induction of metallothionein expression. Metallothionein binds Zn and releases it under stress conditions to exert its antioxidant effect, thereby reducing oxidative stress [104]. Studies have demonstrated the role of Zn as cofactor of SOD, an enzyme that catalyzes the conversion of H2O2 and O2 radicals to less harmful species [105,106]. On the other hand, Zn competes with cell membrane Fe3+ and Cu2+ which catalyze the production of free radicals from lipid peroxides. In this way, the substitution of Fe3+ and Cu2+ with Zn leads to inhibition of free radical production since Zn is catalytically inert [107]. Another enzyme regulated by Zn is glutamate-cysteine ligase, a key enzyme in the synthesis of glutathione. This produces two effects: direct neutralization of free radicals by glutathione, and indirect neutralization of free radicals by Zn as a cofactor of GPx [108,109]. The antioxidant effect of Zn may be beneficial in combating viral infections such as simple herpes and rhinoviruses. Co-administration of Zn and interferon α offers protection against hepatitis C virus infections [110-112]. In studies involving Zn supplementation, a potent antioxidant effect and decreased levels of nitrates, nitrites and DNA were observed [103]. In diseases such as epilepsy, schizophrenia, Alzheimer’s and Parkinson’s, it has been observed that presynaptic neurons release high amounts of Zn which cause neuronal death, NADPH oxidase activation and ROS production [15]. On the other hand, administration of the Zn chelator N,N,N’,N’-tetrakis (2-pyridylmethyl) ethylenediamine attenuated the neurological deficit and cerebral infarction area, increased SOD activity, and decreased serum MDA and interleukin-6 levels [113,114]. Receptive metal transcription factor 1 is a Zndependent transcription factor that stimulates the expression of metallothionein and zinc-1 transporter (ZnT-1) found in cell membranes, and facilitates the exit of excess Zn from the cytosol [115]. Therefore, metallothionein and ZnT-1 help to maintain the intracellular concentration of Zn [115,116]. Zinc transport protein promotes the survival of osteoblastic (MC3T3) cells by preventing cellular aggregation of Zn and apoptosis, and activation of protein tyrosine kinase which catalyzes the phosphorylation of proapoptotic proteins [117,118]. In hyperglycemia, Zn decreases ROS production, thereby reducing oxidative stress, and this improves insulin secretion due to efficient phosphorylation of its receptor, activation of phosphatidylinositol 3-kinase and protein kinase B, which are required for glucose uptake or cellular transport [119,120]. Therefore, it is believed that Zn helps control the action of insulin and serum glucose [120]. A protective effect of Zn consumption is improvement in oxidative stress markers, while its deficiency leads to reduction in the levels of interleukin 2 (IL-2) (a cytokine that responds to microbial infections) and interferon gamma, which is a key cytokine in the adaptation of the immune system to viral and bacterial infections [103]. Thus, Zn is important in inflammatory processes that lead to diseases such as colon cancer, prostate cancer and atherosclerosis.
Zinc and renal failure
Data on the role of Zn in patients with renal failure are scanty. However, it is a known fact that in patients with chronic renal failure, there are disorders in the absorption, metabolism, redistribution and elimination of Zn [121]. Diabetic patients with polyuria usually present with symptoms of Zn deficiency due to its excretion in the urine, which suggest a connection between hyperglycemia, lipid peroxidation and oxidative stress [122]. Similar events are observed in patients with chronic renal failure, possibly due to decrease in mineral intake and low intestinal absorption, and mineral loss during dialysis [122-124]. Elements such as Pb, Fe, Cu and Zn are transported by divalent metal transporter protein which is expressed mainly in the liver. The level of this transporter decreases when some of these elements are ingested, thereby conferring protection on the liver, but it increases in renal cells as a mechanism of excretion which could cause damage to the kidney [125]. Some studies on rats showed that administration of Zn during gestation and lactation may exert a protective antioxidant effect, and prevent renal failure and oxidative stress induced by any other metal, for example As (Table 1) [98].
| Heavy metal |
Oxidative stress effects |
Renal failure effects |
| Mercury |
- Increases the synthesis of H2O2
- Decreases the catalytic activity of GPx
- Increases lipid peroxidation products (MDA and 4-HOA) in mitochondria
- Generation of advanced oxidation protein products
- Increases inflammatory activity
- Damages microtubules and mitochondria
- Interrupts intracellular Ca2+ homeostasis generating an uncontrolled release of Ca2+
- Alters protein phosphorylation
- Generates free –SH groups
- Increases the positive regulation of HO-, O2- and H2O2, and negative regulation of defense enzymes such as SOD, CAT, and GPx, and the functions of GSH
|
- Mercury ions preferentially accumulate in epithelial cells of the renal tubules
- Nephropathy and damage to proximal tubules appear due to exposure to Hg2+
- Exposure to HgCl2 results in tubular, interstitial and glomerular lesions
- Chronic exposure to CH3Hg+ produces fibrotic changes in the glomeruli and deposits of IgG, IgM and C3 in glomerular basement membrane
- Acute tubular necrosis, immunological glomerulonephritis or nephrotic syndrome
- Inorganic Hg, elemental mercury vapors and ingestion of Hg2+ salts result in nephrotic syndrome with proteinuria
|
| Lead |
- ALAD undergoes enolization and autoxidation, which generates O2- anions
- The enolized form of ALAD acts as an electron donor, forming: (i) methemoglobin; (ii) ALAD-radical; and (iii) H2O2
- Radicalized ALAD autoxidizes ROO- and HO-, thus sharpening glutathione oxidation
- Interaction of O2- and H2O2 generates HO-, a powerful oxidant
- Lead ions have high affinity for -SH groups and metal cofactors, thereby decreasing activities of antioxidant enzymes
6. Lead increases the activity of CAT, levels of MDA, GSSG and oxygen free radical, and decreases plasma GSH level
7. Lead displaces selenocysteine group from the active site of glutathione, thereby weakening the enzyme-substrate interaction and down-regulating the activity of GPx
8. An increase in the activity of SOD characterizes exposure to Pb |
- Interferes with mitochondrial function of proximal convoluted tubules
- Decreases the reabsorption of glucose, amino acids and phosphate
- A positive correlation exist between plasma levels of lead (5 g/dl) and urinary excretion of N-acetyl-beta-D-glucosaminidase
- Levels higher than 80 mg/dl causes uric acid retention
- Renal damage may be related to the formation of Schiff bases between proteins and free radicals
|
| Arsenic |
- Arsenic binds to the -SH groups of glutathione, and modifies the redox status (GSH/GSSG)
- Arsenic on binding to lipoate causes the inhibition of and Krebs cycle and interferes with oxidative phosphorylation
- Inhibits the absorption of glucose in cells, and promotes gluconeogenesis and fatty acids oxidation
- Concentration of GSH and activities of G6PDH and GPx are significantly reduced after exposure to Pb
- Arsenic stimulates the generation of high amount of H2O2 via oxidative reactions of Fenton and Haber-Weiss, thereby increasing the production of free radicals
- Increased ROS level alters the functionality of some antioxidant enzymes such as SOD, CAT, GPx and heme oxygenase-1, and concomitant increase in lipid peroxidation
- Cell proliferation and the release of TNF-α are stimulated by ROS
- Oxidative and nitrosative stress reduces the activities of complexes III, IV and V of the respiratory chain, and decreases the level of ATP
|
- kidney is highly sensitive to arsenic toxicity
- Arsenic increases volume and pH of urine
- Decrease in electrolyte levels and increase in Ca2+ excretion
- Increased degeneration of epithelial cells of the proximal convoluted tubule and tubular cylinders, focal mineralization, and podocytes of the Bowman's capsule
- Increased serum levels of urea and creatinine, and histological changes that leads to kidney damage
|
| Zinc |
- Zinc deficiency is associated with an increase in LPO products
- Zinc deficiency induces a conformational change in SOD similar to a mutation, thereby leading to oxidative stress
- Zinc deficiency stimulates the release of pro-inflammatory cytokines, activation of NADPH oxidase and iNOS, thus promoting the production of ROS and RNS
- It competes with the Fe3+ and Cu2+ of the cell membranes, thereby promoting the inhibition of free radicals
- It neutralizes free radicals directly by glutathione and indirectly as a cofactor of GPx
|
- Zinc has a protective antioxidant effect and prevents renal failure and oxidative stress induced by other metals
|
Table 1: Summary of oxidative stress and renal failure effects produced by Hg, Pb, As and Zn deficiency.
Conclusion
This review on heavy metals and kidney toxicity has shown that humans and animals are easily exposed to heavy metals from the environment. On gaining entrance into the body, they compete with essential metals, and exert their nephrotoxic effects by inducing oxidative stress, mitochondrial dysfunction, as well as Ca2+ and ROS-mediated apoptosis. In the kidney, permeability and absorption of epithelial cells are altered by heavy metals (except arsenic), resulting in proteinuria and kidney dysfunction. The antioxidant property of Zn might be potent in mitigating the effect of acute exposure to heavy metals. Zinc deficiency provokes a reduction in antioxidant capacity that contributes to the onset of oxidative stress.
Funding
No financial support was received to prosecute this work.
Conflict of Interest
No conflict of interest is associated with this work.
Ethical Responsibilities
The authors declare that no experiments were conducted on humans or animals to produce this review.
References
- Mishra A, Shukla S (2014) Heavy metal toxicity: a blind evil. J Forensic Res 5: e116.
- Martin S, Griswold W (2009) Human health effects of heavy metals. Environ Sci Technol Briefs Citizens 15: 1-6.
- Flora SJS, Mittal M, Mehta A (2008) Heavy metal induced oxidative stress and its possible reversal by chelation therapy. Indian J Med Res 128: 501-523.
- Morais S, Garcia e Costa F, Pereira ML (2012) Heavy metals and human health. In Oosthuizen J, ed. Environmental Health. Emerging issues and practice. Rikeka, Croatia: IntechOpen 227-246.
- Díaz García JD, Arceo E (2018) Daño renal asociado a metales pesados: trabajo de revisión. Rev Colomb Nefrol 1: 43-53.
- Kumar A, Singh N, Pandey R, Gupta VK, Sharma B (2018) Biochemical and molecular targets of heavy metals and their actions. In: Rai M., Ingle A., Medici S. (eds) Biomedical Applications of Metals. Springer, Cham.
- Singh N, Gupta VK, Kumar A, Sharma B (2017) Synergistic effects of heavy metals and pesticides in living systems. Front Chem 5: 1-9.
- Ayangbenro AS, Babalola OO (2017) A new strategy for heavy metal polluted environments: a review of microbial biosorbents. Int J Environ Res Public Health 14: 94-109.
- Salazar-Lugo R, Lozada U, Rosales M, Astudillo H, Prin J, et al. (2015) Heavy metals and its relationships with biomarkers of oxidative stress in chronic smokers. Rev salud Ambient 15: 88-95.
- Singh N, Kumar A, Gupta VK, Sharma B (2018) Biochemical and molecular bases of lead-induced toxicity in mammalian systems and possible mitigations. Chem Res Toxicol 31: 1009-1021.
- Balmus IM, Ciobica A, Antioch I, Dobrin R, Timofte D (2016) Oxidative stress implications in the affective disorders: main biomarkers, animal models relevance, genetic perspectives, and antioxidant approaches. Oxid Med Cell Longev 2: 1-25.
- Egea J, Fabregat I, Frapart YM, Ghezzi P, Görlach A, et al. (2017) European contribution to the study of ROS: a summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol 13: 94-162.
- Moss DW, Bates TE (2001) Activation of murine microglial cell lines by lipopolysaccharide and interferon- g causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci 13: 529-538.
- Shen H, Liu Z (2006) JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med 40: 928-939.
- Kim Y, Koh J (2002) The role of nadph oxidase and neuronal nitric oxide synthase in zinc-induced poly (ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol 177: 407-418.
- Kupsco A, Schlenk D (2015) Oxidative stress, unfolded protein response, and apoptosis in developmental toxicity. Int Rev Cell Mol Biol 317: 1-66.
- Gupta VK, Singh S, Agrawal A, Siddiqi NJ, Sharm B (2015) Phytochemicals mediated remediation of neurotoxicity induced by heavy metals. Biochem Res Int 534769: 1-9.
- Mori N, Yasutake A, Hirayama K (2007) Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Arch Toxicol 81: 769-776.
- Imai H, Nakagawa Y (2003) Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 34: 145-169.
- Nita M, Grzybowski A (2016) The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev 12: 1-23.
- Farmand F, Ehdaie A, Roberts CK, Sindhu RK (2005) Lead-induced dysregulation of superoxide dismutases, catalase, glutathione peroxidase, and guanylate cyclase. Environ Res 98: 33-39.
- Liu CM, Ma JQ, Sun YZ (2010) Quercetin protects the rat kidney against oxidative stress-mediated DNA damage and apoptosis induced by lead. Environ Toxicol Pharmacol 30: 264-271.
- Kasperczyk A, Machnik G, Dobrakowski M, Sypniewski D, Birkner E, et al. (2012) Gene expression and activity of antioxidant enzymes in the blood cells of workers who were occupationally exposed to lead. Toxicology 301: 79-84.
- Katsuma A, Hinoshita F, Masumoto S, Hagiwara A, Kimura A (2012) Acute renal failure following exposure to metallic mercury. Clin Nephrol 82: 73-76.
- George B, You D, Joy MS, Aleksunes LM (2017) Xenobiotic transporters and kidney injury. Adv Drug Deliv Rev 116: 73-91.
- Obrosova IG, Fathallah L, Liu E, Nourooz-Zadeh J (2003) Early oxidative stress in the diabetic kidney: effect of DL-α-lipoic acid. Free Radic Biol Med 34: 186-195.
- Batuman V, Wedeen RP (2014) The persistence of chronic lead nephropathy. Am J Kidney Dis 64: 1-3.
- Risher JF, Amler SN (2005) Mercury exposure: evaluation and intervention the inappropriate use of chelating agents in the diagnosis and treatment of putative mercury poisoning. Neurotoxicology 26: 691-699.
- Orr SE, Bridges CC (2017) Chronic kidney disease and exposure to nephrotoxic metals. Int J Mol Sci 18: 1039-1073.
- Neeti K, Prakash T (2013) Effects of heavy metal poisoning during pregnancy. Int Res J Environ Sci 2: 88-92.
- Suhartono E, Triawanti, Leksono AS, Djati MS (2014) Oxidative stress and kidney glycation in rats exposed cadmium. Int J Chem Eng Appl 5: 497-501.
- Valko M, Morris H, Cronin M (2005) Metals, toxicity and oxidative stress. Curr Med Chem 12: 1161-1208.
- Patrick L (2002) Mercury toxicity mercury toxicity and antioxidants: part i: role of glutathione and alpha-lipoic acid in the treatment of mercury toxicity. Altern Med Rev 7: 456-471.
- Zalups RK, Koropatnick J (2000) Temporal changes in metallothionein gene transcription in rat kidney and liver: relationship to content of mercury and metallothionein protein 1. J Pharmacol Exp Ther 295: 74-82.
- Dave V, Mullaneya KJ, Goderieb S, Kimelberg HK, Aschnera M (1994) Astrocytes as mediators of methylmercury neurotoxicity: effects on d-aspartate and serotonin uptake. Dev Neurosci 16: 222-231.
- Choi DW (1992) Excitotoxic cell death. J Neurobiol 23: 1261-1276.
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J (1993) NMDA-dependent superoxide production and neurotoxicity. Nature 364: 535-537.
- Sarafian TA (1993) Methyl Mercury Increases Intracellular Ca2+ and Inositol Phosphate Levels in Cultured Cerebellar Granule Neurons. J Neurochem 61: 648-657.
- Farina M, Rocha J, Aschner M (2011) Mechanisms of methylmercury-induced neurotoxicity: evidence from experimental studies. Life Sci 89: 555-563.
- Farina M, Franco JL, Ribas CM, Meotti C, Missau FC, et al. (2005) Protective effects of Polygala paniculata extract against methylmercury-induced neurotoxicity in mice. J Pharm Pharmacol 57: 1503-1508.
- Franco JL, Braga HC, Stringari J, Missau FC, Posser T, et al. (2007) Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: protective effects of quercetin. Chem Res Toxicol 20: 1919-1926.
- Johansson C, Castoldi AF, Onishchenko N, Manzo L, Vahter M, et al. (2007) Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox Res 11: 241-260.
- Tchounwou PB, Ayensu WK, Ninashvili N, Sutton D (2003) Environmental exposure to mercury and its toxicopathologic implications for public health. Environ Toxicol 18: 149-175.
- Roels HA, Hoet P, Lison D, Roels HA, Hoet P (1999) Usefulness of biomarkers of exposure to inorganic mercury, lead, or cadmium in controlling occupational and environmental risks of nephrotoxicity. Ren Fail 21: 251-262.
- Leong CCW, Syed NI, Lorscheider FL (2001) Retrograde degeneration of neurite membrane structural integrity of nervegrowth cones following in vitro exposure to mercury. Neuroreport 12: 733-737.
- Castoldi AF, Coccini T, Ceccatelli S, Manzo L (2001) Neurotoxicity and molecular effects of methylmercury. Brain Res Bull 55: 197-203.
- Guallar E, Sanz-Gallardo M, van’t Veer P, Bode P, Aro A, et al. (2002) Mercury, fish oils, and the risk of myocardial infarction. N Engl J Med 347: 1747-1754.
- Nyland JF, Fairweather D, Shirley DL, Davis SE, Rose NR, et al. (2012) Low-dose inorganic mercury increases severity and frequency of chronic coxsackievirus-induced autoimmune myocarditis in mice. Toxicol Sci 125: 134-143.
- Silva IA, Graber J, Nyland JF, Silbergeld EK (2005) In vitro HgCl 2 exposure of immune cells at different stages of maturation: effects on phenotype and function. Environ Res 98: 341-348.
- Bahn A, Knabe M, Hagos Y, Rödiger M, Godehardt S, et al. (2002) Interaction of the metal chelator 2,3-dimercapto-1- propanesulfonate with the rabbit multispecific organic anion. Mol Pharmacol 62: 1128-1136.
- Farias-Junior P, Teixeira F, Fagundes N, Miranda G, Oliveira Bittencourt L, et al. (2017) Chronic intoxication by methylmercury leads to oxidative damage and cell death in salivary glands of rats. Metallomics 9: 1778-1785.
- Burckhardt BC, Drinkuth B, Menzel C, Steffgen R, Wright SH, et al. (2002) The renal Na+-dependent dicarboxylate transporter, NADC-3, translocates dimethyl- and disulfhydryl-compounds and contributes to renal heavy metal detoxification. J Am Soc Nephrol 3: 2628-2638.
- Zalups RK, Ahmad S (2004) Homocysteine and the renal epithelial transport and toxicity of inorganic mercury: role of basolateral transporter organic anion transporter 1. J Am Soc Nephrol 15: 2023-2031.
- Bridges CC, Joshee L, Zalups RK (2008) MRP2 and the DMPS- and DMSA-mediated elimination of mercury in TR(-) and control rats exposed to thiol S-conjugates of inorganic mercury. Toxicol Sci 105: 211-220.
- Wijkström J, Leiva R, Elinder CG, Leiva S, Trujillo Z, et al. (2013) Clinical and pathological characterization of mesoamerican nephropathy: a new kidney disease in central america. Am J Kidney Dis 62: 908-918.
- Chávez-Gómez N, Cabello-López A, Gopar-Nieto R, Aguilar-Madrid G, Aceves-Valdez M, et al. (2017) Enfermedad renal crónica en México y su relación con los metales pesados. Rev Med Inst Mex Seguro Soc 55: 725-734.
- Kao L, Rusyniak D (2016) Chronic poisoning: trace metals and others. In Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. Philadelphia: Elsevier 92-98.
- Malekirad AA, Oryan S, Fani A, Babapor V, Hashemi M, et al. (2010) Study on clinical and biochemical toxicity biomarkers in a zinc-lead mine workers. Toxicol Ind Health 26: 331-337.
- Chowdhury R, Darrow L, Mcclellan W, Sarnat S, Steenland K (2014) Incident ESRD among participants in a lead surveillance program. Am J Kidney Dis 64: 25-31.
- Rosin A (2009) The long-term consequences of exposure to lead. Isr Med Assoc J 11: 689-694.
- Zhang Z, Wu Z, Feng L, Dong L, Song A (2016) Mg-protoporphyrin IX signals enhance plant’s tolerance to cold stress. Front Plant Sci 7: 1545.
- Somana JS, Uma T (2017) Acute intermittent porphyria. J Evol Res Med Biochem 3: 5-6.
- Said S, Hernandez GT (2015) Environmental exposures, socioeconomics, disparities, and the kidneys. Adv Chronic Kidney Dis 22: 39-45.
- Tandon SK, Singh S, Prasad S, Srivastava S, Siddiqui MKJ (2002) Reversal of lead-induced oxidative stress by chelating agent, antioxidant, or their combination in the rat 1. Environ Res 90: 61-66.
- Ercal N, Treeratphan P, Hammond T, Matthews R, Grannemann N, et al. (1996) In vivo indices of oxidative stress in lead-exposed C57BL/6 mice are reduced by treatment with meso-2,3-dimercaptosuccinic acid or N-acetylcysteine. Free Radic Biol Med 21: 157-161.
- Costa CA, Trivelato GC, Pinto AMP, Bechara EJH (1997) Correlation between plasma 5-aminolevulinic acid concentrations and indicators of oxidative stress in lead-exposed workers. Clin Chem 43: 1196-1202.
- Hermes-Lima M, Pereira B, Bechara E (1991) Are free radicals involved in lead poisoning? Xenobiotica 21: 1085-1090.
- Okamoto OK, Colepicolo P (1998) Response of superoxide dismutase to pollutant metal stress in the marine dinoflagellate gonyaulax polyedra. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 119: 67-73.
- Öktem F, Arslan MK, Dundar B, Delibas N, Gultepe M, et al. (2004) Renal effects and erythrocyte oxidative stress in long-term low-level lead-exposed adolescent workers in auto repair workshops. Arch Toxicol 78: 681-687.
- Ahamed M, Siddiqui MKJ (2007) Low level lead exposure and oxidative stress: Current opinions. Clin Chim Acta 383: 57-64.
- Sun H, Rathinasabapathi B, Wu B, Luo J, Pu L, et al. (2014) Arsenic and selenium toxicity and their interactive effects in humans. Environ Int 69: 148-158.
- Aragón-sulik M, Escolero Fuentes O, Navarro Mendoza S, Ortiz Guzman M (2015) Distribución geográfica de arsénico en acuífero de los Valles Centrales de Oaxaca, Mexico. Ing Hidráulica y Ambient 36: 102-110.
- Aflanie I, Muhyi R, Suhartono E (2015) Effect of heavy metal on malondialdehyde and advanced oxidation protein produtcs concentration: a focus on arsenic, cadmium, and mercury. J Med Bioeng 4: 332-337.
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, et al. (2011) Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol 31: 95-107.
- Sharma B, Singh S, Siddiqi NJ (2014) Biomedical implications of heavy metals induced imbalances in redox systems. Biomed Res Int 2014: 640754.
- Gomez-Caminero A, Howe P, Hughes M, Kenyon E, Lewis DR, et al. (2001) Arsenic and Arsenic Compounds 2nd ed., Chennai: World Health Organization.
- Jomova K, Valko M (2011) Advances in metal-induced oxidative stress and human disease. Toxicology 283: 65-87.
- Maiti S, Chatterjee AK (2001) Effects on levels of glutathione and some related enzymes in tissues after an acute arsenic exposure in rats and their relationship to dietary protein deficiency. Arch Toxicol 75: 531-537.
- Akhtar M, Ashraf M, Anjum A, Javeed A, Sharif A, et al. (2015) Textile industrial effluent induces mutagenicity and oxidative DNA damage and exploits oxidative stress biomarkers in rats. Environ Toxicol Pharmacol 41: 180-186.
- Kodavanti UP, Hauser R, Christiani DC, Meng ZH, Mcgee J, et al. (1998) Pulmonary responses to oil fly ash particles in the rat differ by virtue of their specific soluble metals 1. Toxicol Sci 43: 204-212.
- Ventura JJ, Cogswell P, Flavell RA, Baldwin ASJ, Davis RJ (2004) JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev 18: 2905-2515.
- Jayasumana C, Gunatilake S, Siribaddana S (2015) Simultaneous exposure to multiple heavy metals and glyphosate may contribute to Sri Lankan agricultural nephropathy. BMC Nephrol 16: 103.
- Cohen SM, Yamamoto S, Cano M, Arnold LL (2001) Urothelial cytotoxicity and regeneration induced by dimethylarsinic acid in rats. Toxicol Sci 59: 68-74.
- Mershiba SD, Dassprakash M, Saraswathy S (2013) Protective effect of naringenin on hepatic and renal dysfunction and oxidative stress in arsenic intoxicated rats. Mol Biol Rep 40: 3681-3691.
- Monnier VM, Nemet I, Sell DR, Weiss MF (2010) Transition Metals and Other Forms of Oxidative Protein Damage in Renal Disease. In Miyata T, Eckardt KU, Nangaku M, eds. Studies on Renal Disorders. Oxidative Stress in Applied Basic Research and Clinical Practice. Humana Press, 2010: 25-50.
- Reyes JL, Molina-Jijón E, Rodríguez-Muñoz R, Bautista-García P, Debray-García Y, et al. (2013) Tight junction proteins and oxidative stress in heavy metals-induced nephrotoxicity. Biomed Res Int 2013: 730789.
- Agrawal S, Flora G, Bhatnagar P, Flora SJS (2014) Comparative oxidative stress, metallothionein induction and organ toxicity following chronic exposure to arsenic, lead and mercury in rats. Cell Mol Biol 60: 13-21.
- Shahid F, Rizwan S, Khan W, Khan SA, Naqshbandi A, et al. (2014) Studies on the effect of sodium arsenate on the enzymes of carbohydrate metabolism, brush border membrane, and oxidative stress in the rat kidney. Environ Toxicol Pharmacol 37: 592-599.
- Abdul K, Jayasinghe S, Chandana E, Jayasumana C, De Silva P (2015) Arsenic and human health effects: a review. Environ Toxicol Pharmacol 40: 828-846.
- Dangleben NL, Skibola CF, Smith MT (2013) Arsenic immunotoxicity: a review. Environ Health 12: 73.
- Benters J, Flogel U, Schafer T, Leibfritz D, Hechtenberg S, et al. (1997) Study of the interactions of cadmium and zinc ions with cellular calcium homoeostasis using 19 F-NMR spectroscopy. Biochem J 799: 793-799.
- Prasad AS, Halsted JA, Nadimi M (1961) Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am J Med 31: 532-546.
- Prasad A, Miale A, Farid Z, Sandstead H, Schulert A (1963) Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J Lab Clin Med 61: 537-549.
- Prasad AS (2009) Zinc: role in immunity, oxidative stress and chronic inflammation. Curr Opin Clin Nutr Metab Care 12: 646-652.
- Homma K, Fujisawa T, Tsuburaya N, Yamaguchi N, Kadowaki H, et al. (2013) Article SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol Cell 52: 75-86.
- Oteiza PI (2012) Free radical biology and medicine zinc and the modulation of redox homeostasis. Free Radic Biol Med 53: 1748-1759.
- Weglicki WB, Chmielinska JJ, Kramer JH, Mak IT (2011) Cardiovascular and intestinal responses to oxidative and nitrosative stress during prolonged magnesium deficiency. Am J Med Sci 342: 125-128.
- Nasiry D, Ghabaee Z, Amiri FT, Moghaddam AE, Khalatbary AR, et al. (2017) Administration of zinc against arsenic-induced nephrotoxicity during gestation and lactation in rat model. Soc Diabet Nephrop 6: 74-80.
- Sun Q, Zhong W, Zhang W, Li Q, Sun X, et al. (2015) Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am J Physiol Gastrointest liver Physiol 308: G757-766.
- Taylor KM, Morgan HE, Johnson A, Nicholson RI (2004) Structure-function analysis of HKE4, a member of the new LIV-1 subfamily of zinc transporters. Biochem J 377: 131-139.
- Bray T, Bettger W (1990) The physiological role of zinc as as antioxidant. Free Radic Biol Med 8: 281-291.
- Prasad AS (2008) Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp Gerontol 43: 370-377.
- Prasad AS, Beck FWJ, Bao B, Fitzgerald JT, Snell DC, et al. (2007) Zinc supplementation decreases incidence of infections in the elderly: Effect of zinc on generation of cytokines and oxidative stress. Am J Clin Nutr 85: 837-844.
- Cruz KC, de Oliveira AR, Marreiro Ddo N (2015) Antioxidant role of zinc in diabetes mellitus. World J Diabetes 6: 333-337.
- Lima VB, Sampaio Fde A, Bezerra DL, Moita Neto JM, Marreiro Ddo N (2011) Parameters of glycemic control and their relationship with zinc concentrations in blood and with superoxide dismutase enzyme activity in type 2 diabetes patients. Arq Bras Endocrinol Metabol 55: 701-707.
- Fernandes Cruz J (2011) Uma revisão sobre o zinco. Ensaios e Ciência Ciências Biológicas, Agrárias e da Saúde 15: 207-222.
- Marreiro N, Jayanne K, Beatriz J, Morais S, Beserra B, et al. (2017) Zinc and oxidative stress: current mechanisms. Antioxidants 6: e24.
- Eide DJ (2011) The oxidative stress of zinc deficiency. Metallomics 3: 1124-1129.
- Ha K, Chen Y, Cai J, Sternberg P (2006) Increased glutathione synthesis through an ARE-Nrf2 – dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Investig Ophtalmol Vis Sci 47: 2709-2715.
- Takagi H, Nagamine T, Abe T, Takayama H, Sato K, et al. (2001) Zinc supplementation enhances the response to interferon therapy in patients with chronic hepatitis C. J Viral Hepat 8: 367-371.
- Kümel G, Schrader S, Zentgraf H, Daus H, Brendep M (1990) The mechanism of the antiherpetic activity of zinc sulphate. J Gen Virol 71: 2989-2997.
- Korant B, Kauer J, Butterworth B (1974) Zinc ions inhibit replication of rhinoviruses. Nature 248: 588-590.
- Furuta T, Ohshima C, Matsumura M, Takebayashi N, Hirota E, et al. (2016) Oxidative stress upregulates zinc uptake activity via Zrt/Irt-like protein 1 (ZIP1) in cultured mouse astrocytes. Life Sci 151: 305-312.
- Wang WM, Liu Z, Liu A, Wang Y, Wang H, et al. (2015) The zinc ion chelating agent TPEN attenuates neuronal death/apoptosis caused by hypoxia/ischemia via mediating the pathophysiological cascade including excitotoxicity, oxidative stress, and inflammation. CNS Neurosci Ther 21: 708-717.
- Günther V, Lindert U, Schaffner W (2012) The taste of heavy metals: gene regulation by MTF-1. Biochim Biophys Acta 1823: 1416-1425.
- Sekler I, Sensi SL, Hershfinkel M, Silverman WF (2007) Mechanism and regulation of cellular zinc transport. Mol Med 13: 337-343.
- Liang D, Xiang L, Yang M, Xiuli Z, Guo B, et al. (2013) ZnT7 can protect MC3T3-E1 cells from oxidative stress-induced apoptosis via PI3K/Akt and MAPK/ERK signaling pathways. Cell Signal 25: 1126-1135.
- Sun Q, Li Q, Zhong W, Zhang J, Sun X, et al. (2014) Dysregulation of hepatic zinc transporters in a mouse model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 307: G313-322.
- Ranasinghe P, Pigera S, Galappatthy P, Katulanda P, Constantine GR (2015) Zinc and diabetes mellitus: understanding molecular mechanisms and clinical implications. DARU J Pharm Sci 23: 44.
- Vardatsikos G, Pandey NR, Srivastava AK (2013) Insulino-mimetic and anti-diabetic effects of zinc. J Inorg Biochem 120: 8-17.
- Yonova D, Vazelov E, Tzatchev K (2012) Zinc status in patients with chronic renal failure on conservative and peritoneal dialysis treatment. Hippokratia 16: 356-359.
- Tonelli M, Wiebe N, Hemmelgarn B, Klarenbach S, Field C, et al. (2009) Trace elements in hemodialysis patients: a systematic review and meta-analysis. BMC Med 7: 25.
- Noleto Magalhães RC, Guedes Borges de Araujo C, Batista De Sousa Lima V, Machado Moita Neto J, do Nascimento Nogueira N, et al. (2011) Nutritional status of zinc and activity superoxide dismutase in chronic renal patients undergoing hemodialysis. Nutr Hosp 26: 1456-1461.
- Habib SA, Saad EA, Elsharkawy AA, Attia ZR (2015) Pro-inflammatory adipocytokines, oxidative stress, insulin, Zn and Cu: interrelations with obesity in Egyptian non-diabetic obese children and adolescents. Adv Med Sci 60: 179-185.
- Ruíz Ramírez DC, Alcaráz Contreras Y, Martínez Alfaro M (2017) Estudio de la toxicidad por plomo y la función de algunas proteínas. Jovenes en la Cienc 3: 252-256.