Keywords
Nonalcoholic fatty liver disease (NAFLD); Epigenetics; DNA methylation; Liver; Steatosis; Histone modifications
Abbreviations
NAFLD: Non-Alcoholic Fatty Liver Disease; HCC: Hepatocellular Carcinoma; CpG: Cytosine-Phospho-Guanine; DNMT: DNA Methyltransferase; HATs: Histone Acetyltransferases; HDAC: Histone Deacetylase; ncRNA: Non-Coding RNA
Introduction
Non-alcoholic fatty liver disease (NAFLD) is characterized by the excessive accumulation of fat, particularly triglycerides, in the hepatocytes without alcohol consumption, which can lead to liver cirrhosis and hepatocellular carcinoma (HCC) [1,2]. NAFLD has been emerge as a most common cause of liver disease in Western countries and includes nonalcoholic steatohepatitis (NASH) [3,4]. The prevalence of NAFLD in the United States (U.S.) has increased from 18% in 1988– 1991 to 29% in 1999–2000 to 31% in 2011–2012 [5]. Estimates of NAFLD prevalence for adults in Western countries is 80%-90% in obese adults, 30%- 50% in patients with diabetes and up to 90% in patients with hyperlipidemia [6]. In light of the rapid rise in cirrhosis as well as hepatocellular carcinoma resulting from NAFLD, studying the pathogenesis of NAFLD represents an urgent task.
It has been reported that lifestyle aspects such as physical inactivity, overnutrition, metabolic disorder like insulin resistance and weight gain can influence the development and progression of NAFLD via epigenetic mechanisms [7,8]. The role of epigenetic factors are progressively being identified as a crucial link between environmental exposures, genetic determinants, and disease risk, which can influence gene expression without changing the DNA sequence, offers a new perspective on the pathogenesis of NAFLD [7-10]. The epigenetic modulation of gene expression, which can induce phenotypic changes, may occur in response to environmental cues in three major systems namely (1) a modification of the DNA nucleotides (DNA methylation), (2) modifications of histones, key protein components of nucleosomes, which regulate the chromatin compactness and accessibility (histone tail modifications), and (3) non-coding RNA (ncRNA) mediated gene silencing (Figure 1) [11-14].
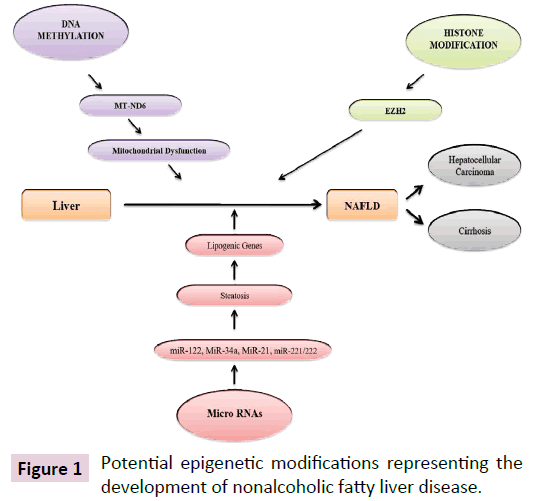
Figure 1: Potential epigenetic modifications representing the development of nonalcoholic fatty liver disease.
All these three major epigenetic mechanisms control the chromatin structure, modifications, and the initiation of transcription in such a way that modifies the accessibility of genes to transcription factors and their cofactors that control the rate at which a gene is vigorously transcribed [11-14]. Hence, it is not surprising that epigenetics has become a research area of considerable interest, relating alterations in chromatin states to the cellular phenotype and, in turn, the functioning of an organ. Several reports have been examined the effect of epigenetic variations on both liver function and disease states [15,16]. This review focuses on the epigenetic mechanisms engaged in the pathogenesis of hepatic fibrosis and also describes current epigenetic approaches and strategies for the treatment of NAFLD.
Epigenetic Mechanism in NAFLD
Modulation of DNA methylation in NAFLD
DNA methylation, a biochemical modification of cytosine in DNA with a methyl group (one-carbon moiety), is the earliest discovery of epigenetic regulation of gene expression [17-19]. The DNA methylation reaction is catalyzed by DNA methyltransferases (DNMTs), which requires the addition of a methyl group to cytosine with guanine as the next nucleotide, known as CpG sites [20]. The grouping of CpG dinucleotides (typically denoted as CpG islands and CpG island shores) is generally occur with higher frequency at the promoter regions of the genes than at other DNA sites [21]. Hypermethylation of CpG islands is usually linked with gene silencing, while hypermethylation of heterochromatin area effects genomic stability [22,23]. Irregular methylation patterns of genomic DNA known as one of the key epigenetic modifications that can induce aberrant gene expression in NAFLD. Moreover, it is also known that epigenetic modifications in mitochondrial DNA methylation may arise during the development of NAFLD [24]. There are three isoforms of DNMT are present in human (DNMT1, DNMT3A and DNMT3B) [25]. In a mouse model, the hepatic epigenetic phenotype predetermined its vulnerability to hepatic steatosis, which was linked with variations in DNMT1 and DNMT3A expression in the liver [26]. In human clinical study, the methylated/unmethylated DNA ratio of mitochondrially encoded NADH dehydrogenase (MT-ND6) was remarkably allied with NAFLD activity score, a tool to quantity variations in NAFLD during therapeutic trials using feature-based scoring of histologic lesions [24,26]. It has been reported that differentially methylated genes might distinguish patients with advanced NASH from simple steatosis [27]. A recent epigenetic clinical study suggested that in advanced NAFLD, a huge number of tissue repair genes were hypomethylated in the liver, whereas genes for metabolic pathways including one-carbon metabolism were hyper methylated. In a mild versus severe NAFLD cohort, DNA methylation has demonstrated noteworthy alterations in several CpG sites within fibrosis-linked genes [28]. It seems that the DNA methylation status at specific CpGs could be beneficial for predicting the progression of NAFLD to NASH fibrosis. Moreover, DNA methylation profiles in genes linked with lipid homeostasis, fibrosis, and carcinogenesis could be supportive to find the etiologic function of DNA methylation in the development of NAFLD [28].
Impairment in the regulation of histone code in NAFLD
Post-translational alterations of histones, which comprise of acetylation, methylation, ribosylation, ubiquitination, phosphorylation and sumoylation of histone tails, constitute a key factor of chromatin compactness and accessibility [29]. Between them, the acetylation of lysine residues at the N-terminus of histone tails has been most widely explored [30]. Histone acetylation is catalyzed by histone acetyltransferases (HATs), while histone deacetylation is catalyzed by histone deacetylases (HDACs) [31]. The histone acetylation status is regulated by the balance between HAT and HDAC. It has been reported that histone acetylation affects the gene expression profiles in NAFLD [32]. Naturally, abnormal histone alterations induce the progression of type 2 diabetes mellitus along with insulin resistance and subsequently develop NAFLD [33]. p300, a member ofthe HAT family, is a key transcriptional controller that is involved in the NF-kB dependent inflammatory pathways [34]. Hyperglycemia induced activation of p300 enhances ChREBP transcriptional activity and promotes the progression of NAFLD via the activation of lipogenic genes by histone and non-histone protein acetylation [32]. Therefore, suppression of hepatic p300 activity may be useful for the treatment of hepatic steatosis and that specific p300 blockers may be a potential therapeutic strategy for the intervention of NAFLD [35]. There are four families of HDACs (class I, IIa, IIb and IV), which vary in structure, enzymatic function and subcellular localization. In addition, there is another group of deacetylases, the sirtuins, which are denoted to as class III HDACs. Various HDACs also participate in a fundamental function in the development of NAFLD. HDAC3, a member of human class I HDACs, controls the circadian rhythm of hepatic lipogenesis [36]. In vivo study disturbance of the circadian rhythm orchestrated by HDAC3 deranged the hepatic lipid metabolism and subsequently caused obesity and insulin resistance [37]. HDAC3 also was found to combine to the mouse liver genome in a circadian pattern and controlled the expression of genes for hepatic lipid homeostasis, which caused liver steatosis when disrupted [36,37]. These studies suggest that genes activated in the livers of patients with NAFLD strongly correlate with histone modification marks.
Small non-coding RNAs (miRNAs) as epigenetic mediator in NAFLD
MicroRNAs (miRNAs) are small non-coding, endogenous, single stranded RNAs typically made up of 21-25 nucleotides that control gene expression via suppression or degradation of targeted mRNAs [38]. MiRNAs are identified to affect the phenotypic expression by fine-tuning of gene expression and recently have been proposed as potential biomarkers and attractive therapeutic targets for NAFLD [38]. After amalgamation into the RNA-induced silencing complex (RISC), miRNAs can target mRNAs via complementary base pairing, thus stipulating the posttranscriptional suppression of targeted protein-coding genes, either by transcript disruption or by translational suppression [39]. MiRNAs can affect a broad range of biological processes like glucose as well as lipid metabolisms, which have been recognized to be epigenetically decontrolled in NAFLD [40]. The potential roles of miRs in both physiological homeostasis and pathogenesis have been highlighted in the liver and their dysregulation to epigenetic or environmental cues may contribute to the development and progression of NAFLD and metabolic syndrome [41]. One of the miRNAs that has been described to be linked with lipid metabolism and homeostasis is miR-122, the most abundant miRNA in adult human liver, embracing 70% of the total miRNA expressed in the liver [42]. MiR-34a, which is extremely expressed in patients with NAFLD and insulin resistance, is one of the most lipid-responsive hepatic miRNAs and the hepatic expression levels of miR-34a are associated with the severity of NASH [43]. MiR-21, one of the first miRNAs recognized as an oncomir, is over-expressed in the liver of patients with NAFLD and the severity of NASH is completely linked with the expression of hepatic miR-21 [44]. Suppression of miR-21 restored the expression of HMG-box transcription factor 1 (HBP1), a transcriptional activator of the tumor suppressor p53, demonstrating a mechanism by which miR-21 can affect p53 governing hepatic lipogenesis and HCC progression from NAFLD [44]. The over-expression of miR-221/222 has been identified in the livers of NAFLD patients in studies of the ramification of hepatic stellate cell activation and fibrosis [45]. Mice upregulating miR-221/222, which targets the gene p27 governing cell cycle and PTEN, also exhibited HCC development from NAFLD [46]. In NAFLD patients, miR-122, miR-192, miR-19a, miR-19b, miR-125 and miR-375 levels in the serum were found at least two-fold greater than the healthy controls, while liver tissue expression of these miRNAs was simultaneously lesser in the NAFLD patients [41]. Taken together, down-regulation of miRNAs could be a potential therapeutic approach against NAFLD.
Conclusion and Future Prospectives
Recent reports describe the governing effect that epigenetic alterations exert on NAFLD process; yet, despite many published data, epigenetics are far from being cleared. Next-generation sequencing, along with advances in molecular technologies such as CRISPR/Cas9 are widely used to discover innovative gene monitoring pathways and distinct epigenetic mechanisms, which will open a new chapter in the study of NAFLD as well as HCC development in NASH. Among epigenetic mechanisms, miRNAs are receiving the most attention, because their disturbances present potential prognostic and diagnostic, and the ability to be therapeutic targets. Impending epigenetic studies are required to enhance information of the role that epigenetics mechanisms could participate in regulating extremely violent phenotypes of NAFLD. This could prompt to disease amalgamation, from simple steatosis to non-alcoholic steatohepatitis, in order to target therapies, providing new tracks in NAFLD pathogenesis.
References
- Angulo P (2002) Non-alcoholic fatty liver disease. N Engl J Med 346: 1221-1231.
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, et al. (2005) The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 129: 113-121.
- Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, et al. (2011) Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 140: 124-131.
- Ajmera VH, Terrault NA, Harrison SA (2017) Is moderate alcohol use in nonalcoholic fatty liver disease good or bad? A critical review. Hepatology 65: 2090-2099.
- Ruhl CE, Everhart JE (2015) Fatty liver indices in the multiethnic United States National Health and Nutrition Examination Survey. Aliment Pharmacol Ther 41: 65-76.
- Le MH, Devaki P, Ha NB, Jun DW, Te HS, et al. (2017) Prevalence of non-alcoholic fatty liver disease and risk factors for advanced fibrosis and mortality in the United States. PLoS One 12: e0173499.
- Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, et al. (2010) Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology 52: 1992-2000.
- Lee JH, Friso S, Choi SW (2014) Epigenetic mechanisms underlying the link between non-alcoholic fatty liver diseases and nutrition. Nutrients 6: 3303-3325.
- Slomko H, Heo HJ, Einstein FH (2012) Minireview: Epigenetics of obesity and diabetes in humans. Endocrinology 153: 1025-1030.
- Zimmer V, Lammert F (2011) Genetics and epigenetics in the fibrogenic evolution of chronic liver diseases. Best Pract Res Clin Gastroenterol 25: 269-280.
- de Conti A, Ortega JF, Tryndyak V, Dreval K, Moreno FS, et al. (2017) MicroRNA deregulation in nonalcoholic steatohepatitis-associated liver carcinogenesis. Oncotarget 8: 88517-88528.
- Mehra M, Chauhan R (2017) Long non-coding RNAs as a key player in hepatocellular carcinoma. Biomark Cancer 9: 1179299X17737301.
- Vickers MH (2014) Early life nutrition, epigenetics and programming of later life disease. Nutrients 6: 2165-2178.
- Yokoyama M, Chiba T, Zen Y, Oshima M, Kusakabe Y, et al. (2017) Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget 8: 21315-21326.
- Eslam M, Valenti L, Romeo S (2017) Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol pii: S0168-8278(17)32282-1.
- Wilson CL, Mann DA, Borthwick LA (2017) Epigenetic reprogramming in liver fibrosis and cancer. Adv Drug Deliv Rev 121: 124-132.
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, et al. (2010) Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 6: e1000952.
- Ding CJ, Liang LX, Diao S, Su XH, Zhang BY (2018) Genome-wide analysis of day/night DNA methylation differences in Populus nigra. PLoS One 13: e0190299.
- Maschietto M, Rodrigues TC, Kashiwabara AY, de Araujo ÉSS, Marques Aguiar TF, et al. (2016) DNA methylation landscape of hepatoblastomas reveals arrest at early stages of liver differentiation and cancer-related alterations. Oncotarget 8: 97871-97889.
- Portela A, Liz J, Nogales V, Setién F, Villanueva A, et al. (2013) DNA methylation determines nucleosome occupancy in the 5'-CpG islands of tumor suppressor genes. Oncogene 32: 5421-5428.
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature 518: 317-330.
- de Mello VD, Matte A, Perfilyev A, Männistö V, Rönn T, et al. (2017) Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 12: 287-295.
- Chen WJ, Cai B, Chen HT, Cao CY, Du YL, et al. (2016) The role of ADIPOQ methylation in curcumin-administrated experimental nonalcoholic fatty liver disease. J Dig Dis 17: 829-836.
- Pirola CJ, Gianotti TF, Burgueño AL, Rey-Funes M, Loidl CF, et al. (2006) Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 62: 1356-1363.
- Klose RJ, Bird AP (2006) Genomic DNA methylation: The mark and its mediators. Trends Biochem Sci 31: 89-97.
- Pogribny IP, Tryndyak VP, Bagnyukova TV, Melnyk S, Montgomery B, et al. (2009) Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J Hepatol 51: 176-186.
- Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, et al. (2013) Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 145: 1076-1087.
- Zeybel M, Hardy T, Robinson SM, Fox C, Anstee QM, et al. (2015) Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin Epigenetics 7: 25.
- Chen ZJ, Pikaard CS (1997) Epigenetic silencing of RNA polymerase I transcription: A role for DNA methylation and histone modification in nucleolar dominance. Genes Dev 11: 2124-2136.
- Hardy T, Mann DA (2016) Epigenetics in liver disease: From biology to therapeutics. Gut 65: 1895-1905.
- Xie M, Kong Y, Tan W, May H, Battiprolu PK, et al. (2014) Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129: 1139-1151.
- Tian Y, Wong VW, Chan HL, Cheng AS (2013) Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin Cancer Biol 23: 471-482.
- Ling C, Groop L (2009) Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 58: 2718-2725.
- Chan HM, La Thangue NB (2001) p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114: 2363-2373.
- Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, (2010) Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest 120: 4316-4331.
- Gallego-Durán R, Romero-Gómez M (2015) Epigenetic mechanisms in non-alcoholic fatty liver disease: An emerging field. World J Hepatol 7: 2497-2502.
- Feng D, Liu T, Sun Z, Bugge A, Mullican SE, et al. (2011) A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331: 1315-1319.
- He Y, Huang C, Zhang SP, Sun X, Long XR, et al. (2012) The potential of microRNAs in liver fibrosis. Cell Signal 24: 2268-2272.
- Gao L, Jiang F (2016) MicroRNA (miRNA) Profiling. Methods Mol Biol 1381: 151-161.
- Awazawa M, Gabel P, Tsaousidou E, Nolte H, Krüger M, et al. (2017) A microRNA screen reveals that elevated hepatic ectodysplasin A expression contributes to obesity-induced insulin resistance in skeletal muscle. Nat Med 23: 1466-1473.
- Pirola CJ, Fernández Gianotti T, Castaño GO, Mallardi P, San Martino J, et al. (2015) Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 64: 800-812.
- Fan B, Sutandy FX, Syu GD, Middleton S, Yi G, et al. (2015) Heterogeneous Ribonucleoprotein K (hnRNP K) Binds miR-122, a Mature Liver-Specific MicroRNA Required for Hepatitis C Virus Replication. Mol Cell Proteomics 14: 2878-2886.
- Kong L, Zhu J, Han W, Jiang X, Xu M, et al. (2011) Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol 48: 61-69.
- Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, et al. (2015) Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 308: G298-G312.
- Ogawa T, Enomoto M, Fujii H, Sekiya Y, Yoshizato K, et al. (2012) MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut 61: 1600-1609.
- Callegari E, Elamin BK, Giannone F, Milazzo M, Altavilla G, et al. (2012) Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology 56: 1025-1033.

