Keywords
Epigenetics; DNA remodeling; Chronic regional pain syndrome; Cellular function
Introduction
Our understanding of pain has increased significantly over the past few decades. A recent shift in scientific emphasis that treats chronic pain as a disease rather than as a symptom has led to more basic and translational research initiatives. It is now widely accepted that there are molecular, physiologic, and perceptual differences between acute and chronic pain [1,2]. The presentation of a noxious stimulus to an organism provokes molecular changes in pain fibers that affect nerve sensitivity and excitability. Ultimately, this may determine whether a pain fiber will carry acute or chronic nociceptive information [3]. There are many phases of neural pain recognition and transmission at the cellular level, each contributing to membranous or nuclear change mediated by upregulation of protein translation and membrane-receptor coupling [4-8]. The initial stages take seconds to complete and the latter stages may take as long as several weeks. An acute nociceptive insult can precipitate chronic pain, as may be the case with fibromyalgia and Chronic Regional Pain Syndrome (CRPS) [9-12]. There has been a growing interest in examining how cellular morphology and function can be modulated with such speed, given our understanding that protein production via coupling transcription and translation takes significantly longer to occur.
We know that cellular function is intimately linked to genetic processes. Therefore, it logically follows that a change in cellular performance must be accompanied by a change in genetic activity. Subjects who suffer from chronic pain may have different genetic architecture and activity in neurologic pain transmitting locations as compared to subjects who do not suffer from chronic pain [13-15]. To date, no one has been able to describe how a painful experience can cause persistent genetic alterations that affect cellular processing and, consequently, the subjective experience of pain. However, we have recently come to learn that the nervous system is remarkably plastic and genetically modifiable in response to environmental cues. Here, we intend to describe that the link between acute and chronic pain lies within environmentally-induced, epigenetic influences that alter genomic expression and cellular function [16]. Many investigators have recently begun to elucidate the roles of epigenetic processes, in neuronal homeostasis and cellular dynamic [17,18]. We suggest that these molecular events may be responsible for synaptic, axonal, and neural circuit and network alterations in association with dynamic gene-environmental interactions during a response to a nociceptive stimulus and, hence, for modulation of chronic pain. We will examine how these factors can influence nociception and suggest a novel approach toward understanding and treating pain.
Epigenetic Mechanisms Underlying Pain
Epigenetic mechanisms
The molecular networks identified in pain transmission and modulation elucidate our immediate response to environmental pain cues, but we do not have a philosophical template to explain the conversion of those transient signals to the sustained neural potentials seen in chronic pain. Here, we will attempt to explain how the nociceptive system can be epigenetically transformed into a disregulated network which propagates pain signals without a stimulus.
Epigenetics refers to a newly defined multi-layered genomewide regulatory system that orchestrates dynamic and selective changes in gene expression and function. It also refers to the modulation of integrated gene networks mediated by molecular mechanisms that include DNA methylation; histone posttranslational modifications (PTMs), nucleosome repositioning and chromatin remodeling; non-coding RNAs and RNA editing.
Epigenetic changes during neuropathic inflammation and pain
Many biophysiological studies have implicated inflammatory mediators in pain transmission. It is becoming increasingly clear that the nature of somatic, visceral, nociceptive, and neuropathic pain has an underlying inflammatory component. The dorsal horn and peripheral nociceptive circuitry display immunologic activity after damaging and non-damaging painful stimuli [19-24]. Thus, the study of inflammation may be the key to understanding how to manage all types of pain effectively.
A myriad of epigenetic processes have been observed in inflammation, including histone modification, allelic silencing, and other associated regulatory mechanisms [24]. Inflammation and pain represent an organism’s response to tissue damage and are inextricably linked. However, in chronic pain syndromes, pain persists while clinical signs of inflammation have resolved. We propose that epigenetics may be a plausible explanation for this phenomenon, providing a conceptual and molecular link between an environmental insult to the organism and the perpetual sensation of pain, sustained by changes at the level of the genome. Hence, epigenetics may elucidate individual variations in response to nociceptive stimuli.
Direct neural insult results in extrusion of axodendritic contents into the extracellular fluid, the recruitment of inflammatory cells, and the production and release of ions, amines (histamines), kinins (bradykinin), prostanoids (prostaglandin E2), purines (ATP), nitrous oxide, cytokines (IL-1, TNF-alpha, IL-6), and growth factors (leukemia inhibitory factor, nerve growth factor-NGF) [22,23,25]. These agents, via direct activation of receptors or via early post-translational changes, act upon the peripheral terminals of nociceptors to lower the excitation potential within seconds. It is either the transducer molecule or sodium channels in the terminal that are affected. Many inflammatory mediators activate protein kinase A or C, both of which phosphorylate the TTXr sodium channel and also up-regulate the heat sensitive receptor, VR1 [26,27]. This phenomenon is termed peripheral sensitization and is associated with immediate post-traumatic hyperalgesia. Intradiscal administration of Etanercept, a TNF-alpha inhibitor, has been shown to affect pain receptor modulation via CGRP levels in mouse dorsal horn neurons. However, animal models seem to be consistent with human studies. TNF-alpha, IL-1, CGRP, and IL-6 levels have been been demonstrated in patients within injured lumbar discs.
Inflammation can magnify and potentiate pain in two ways. Inflammation is associated with an increase in systemic NGF and brain derived neurotrophic factor (BDNF) levels; this may serve as the key signal for transcriptional activation by CREB within the dorsal root ganglion (DRG) and in the dorsal horn [2]. In addition, there are transcription-dependent molecular alterations that mediate long-term change in the DRG and dorsal horn that produce phenotypic and functional switching of high threshold C-fibers to low threshold A-beta fibers [2,27-30]. Local antagonism of Tropomyosin-related Kinase A receptor (TrKA), a receptor associated in the NGF pathway which mediates receptor sensitivity, has been demonstrated to decrease pain in human subjects [31]. Furthermore, an increase in the expression of TrkB and substance P in the DRG and in the dorsal horn is a prominent feature of inflammatory pain.
Second, the local and systemic growth factors NGF and BDNF are produced during an inflammatory reaction and mediate ligandreceptor internalization within traumatized tissue. Facilitated by G-proteins Ras, Raf, and MAP kinase kinase (MAPKK), these internalized and activated receptor complexes travel in an anterograde direction to the DRG with eventual activation and phosphorylation of MAPK. Following phosphorylation, a component of activated MAPK translocates to the nucleus, where further phosphorylation events activate transcription factors including: c-Myc, Elk-1, c-Fos, and c-Jun [32]. These processes promote dynamic nuclear remodeling and the subsequent coupling of transcription to post-transcriptional processing and translation to generate specific classes of protein products to prolong potentiation and decrease the threshold for receptor activation, the molecular underpinnings of clinical allodynia (Figures 1A-C and 2A-D).
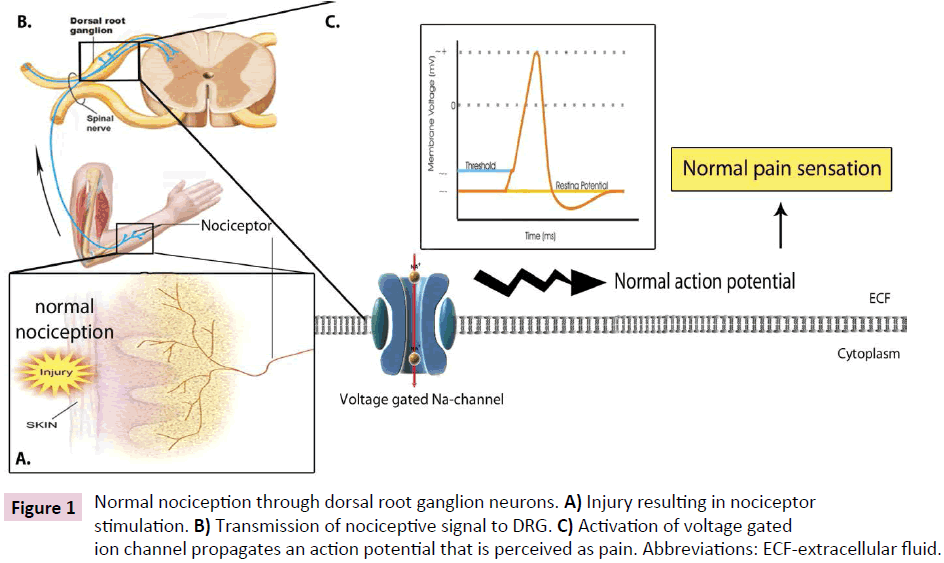
Figure 1: Normal nociception through dorsal root ganglion neurons. A) Injury resulting in nociceptor stimulation. B) Transmission of nociceptive signal to DRG. C) Activation of voltage gated ion channel propagates an action potential that is perceived as pain. Abbreviations: ECF-extracellular fluid.
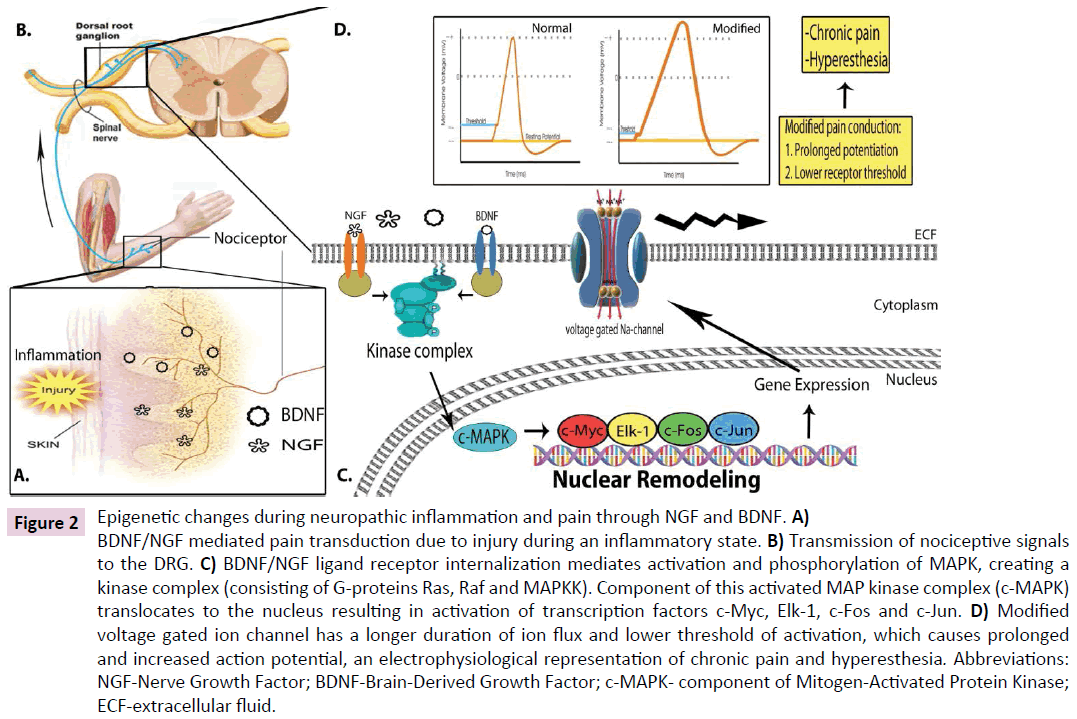
Figure 2: Epigenetic changes during neuropathic inflammation and pain through NGF and BDNF. A) BDNF/NGF mediated pain transduction due to injury during an inflammatory state. B) Transmission of nociceptive signals to the DRG. C) BDNF/NGF ligand receptor internalization mediates activation and phosphorylation of MAPK, creating a kinase complex (consisting of G-proteins Ras, Raf and MAPKK). Component of this activated MAP kinase complex (c-MAPK) translocates to the nucleus resulting in activation of transcription factors c-Myc, Elk-1, c-Fos and c-Jun. D) Modified voltage gated ion channel has a longer duration of ion flux and lower threshold of activation, which causes prolonged and increased action potential, an electrophysiological representation of chronic pain and hyperesthesia. Abbreviations: NGF-Nerve Growth Factor; BDNF-Brain-Derived Growth Factor; c-MAPK- component of Mitogen-Activated Protein Kinase; ECF-extracellular fluid.
DNA Methylation
DNA methylation is a chemical modification of DNA that can be inherited and subsequently removed, altering the expression of DNA without modifying the DNA sequence. DNA methylation involves the addition of a methyl group to the 5’ carbon moiety of the cytosine pyrimidine ring. Preferential sites of DNA methylation are DNA strands with a high concentration of cytocine and guanine (CpG islands) repeats in all tissues. Alterations in the level of DNA methylation are a known mechanism for finetuning gene expression in response to changing environmental conditions [33].
DNA methylation of the opiate receptor
Opiate receptors are a group of G-protein coupled receptors that bind opioids as ligands and are found throughout primate organ systems [34]. Three subtypes, mu (MOR), kappa (KOR), and delta (DOR) are the most studied in relation to pain. Mu and kappa opioid receptors are found in the brain and dorsal horn, whereas delta opioid receptors are located predominantly in the brain [35-38]. In response to painful stimuli, the organism generates endorphins, which upon binding opioid receptors, modulates glutaminergic, GABAergic, serotonergic, cholinergic, and substance P releasing nerves by initiating presynaptic adenylyl cyclase/MAPK cascades. The clinical effects of ligand binding can be analgesia, euphoria, impulsivity, gastrointestinal/renal dysmotility, confusion, or even increased pain. Furthermore, the response to ligand binding is variable; some individuals may experience all of the aforementioned symptoms, others will experience none. This variability is thought to be related to individual receptor subtype predisposition. For example, patients who experience euphoria, impulsivity, and addiction with exogenous opioid treatment may have relatively more MOR (with greater endorphin binding affinity) than KOR or DOR [38]. Transcriptional regulation of the three major opioid receptors have revealed an underlying epigenetic regulation that may explain the prevalence of opioid receptor subtypes [37]. In undifferentiated embryonic neurons, the chromatin of endogenous KOR exists in an unfolded state to allow the transcription factor Sp1 to freely bind to the KOR promoter region. However, the addition of retinoic acid (RA) to the culture condensed the KOR chromosome to inhibit transcription binding. Addition of nerve growth factor (NGF) altered the methylated epigenetic marks on the KOR promoter from silent (H3K9-me2) to active (H3K4-me2) for 6 to 8 days; this activation correlated with sustained and increased binding affinity for the KOR promoter transcription factor AP2.
This evidence suggests that our pain perception and idiosyncratic behavioral responses to pain are epigenetically preprogrammed in utero via opiate receptor differentiation, transacted by neurotrophic growth factor mediated DNA methylation. However, it also presents the possibility for epigenetically induced variability in adult differentiated pain cells. It is known that some adult differentiated cells have the capacity to undergo adaptive and pathological epigenetic change [38,39]. The cell types subjected to such change have an inherent ability to direct DNA transcription and protein production in order to respond rapidly to environmental stressors. Immune cells are known to undergo this type of transformation because of their ability to promptly alter surface proteins to prepare for attack on a variety of pathogens, but we have seen that adult nociceptors also demonstrate a remarkable capacity to react, adapt, and modulate environmental stimuli [40-42]. Thus, we infer that adult differentiated pain neurons may have an intrinsic mechanism for plasticity, perhaps by the same epigenetic mechanisms that developed in utero.
DNA Methylation of the Reelin Gene and Spinal Cord Pain Processing
Reelin is a developmental extracellular matrix protein involved in CNS neuroblast migration, cortical and cerebellar GABAergic neuronal functioning, and memory formation. It is deregulated in numerous neuropsychiatric conditions in which pain is a common symptom [33,42]. It is responsible for nervous system glutamate receptor maturation, axonal vesicular transport, and microtubule formation [33,44]. Reelin has been identified in the dorsal horn laminae I, II and V pain modulating centers, and linked to neuronal function and plasticity, abnormal dorsal horn pain processing, and opiate analgesia sensitivity [44,45].
DNA methylation of the Reelin gene promoter sequence is the primary mode of transcriptional control. Within the dorsal horn, Reelin has been suggested to mediate the dynamic interlaminar sprouting and synaptic cross-talk known to occur hours after a noxious event, whereas impaired Reelin directed cellular positioning and plasticity has demonstrated impaired nociception [44-51]. Therefore, defective DNA methylation of the Reelin promoter can alter pain signaling and intensity via a Reelin-NMDA coupling mechanism.
Dopamine Metabolism, DNA Methylation, and Pain
Dopamine is a major pain modulating neurotransmitter which acts on the descending tracts of the spinal cord and brain to suppress pain [33,52-55]. Positron emission tomography and molecular genetic studies performed on healthy volunteers and patients suffering with chronic pain have shown that dopamine (DA) processing is impaired in the thalamus, periaqueductal gray (PAG), basal ganglia, insular cortex, and spinal cord [56,57]. Chronic pain has been documented in many painful disorders in which DA is thought to be the main influencing factor such as “burning mouth syndrome,” fibromyalgia, and restless leg syndrome [53,55-58]. Dopaminergic drugs such as antipsychotics, anti-parkinsonian drugs, atypical antidepressants, and antipsychotics have also been effectively used to modify pain perception.
Recently, DA metabolism has been proposed as an epigenetically modifiable phenomenon characterized by the DNA methylation of promoter regions of genes involved in dopaminergic metabolism [33]. It is well documented that patients with schizophrenia and bipolar disorder have altered DA processing. It is also known that these subgroups have a higher incidence of chronic pain complaints than patients without these disorders [33,59,60]. In a recent post-mortem study of patients with schizophrenia and bipolar disorder, hypomethylation of the COMT gene (the product of which lowers DA concentration in synapses) was seen relative to controls. DR1 and DR2 promoters were correspondingly hypermethylated and exhibited decreased expression of DR1 and DR2. Patients with the COMT gene polymorphism, COMTVal158Met, demonstrated a genetic predisposition for COMT hypomethylation and, thus, was the suggested reason for DR1 and DR2 hypermethylation. These findings demonstrate that our threshold for pain may be regulated by a DNA methylating epigenetic mechanism and further suggest that a genetic predisposition for epigenetic modification of genes involved in DA metabolism may alter pain perception.
BDNF DNA Methylation
BDNF is critical for neuronal survival, differentiation, and modulation of synaptic strength. It is the common denominator in inflammatory pain mechanisms and is controlled through epigenetic modulation. The BDNF gene is complex with multiple promoter sites allowing for differential splicing. Thus, BDNF mRNA can exist in many different isoforms. At least six BDNF mRNA isoforms have been described, each having its own corresponding mRNA. Differential BDNF expression can be induced by environmental factors. BDNF isoforms via mRNA intermediates I, IV, and VI have been associated with modifications in DNA methylation in mouse hippocampal neurons when subjects were exposed to painful stimuli. Concomitant infusion of zebularine (a DNA methyltransferase inhibitor) increased total BDNF expression, with selective elevations in type I, IV, and VI transcripts from the basal state demonstrating that pain may downregulate BDNF DNA methylation and increase selective production of BDNF isoforms. In addition, hippocampal NMDA receptor inhibition prevented altered BDNF gene expression and memory formation of pain responses [61]. This suggests that an acutely painful NMDA transduction mediated experience may result in protracted and altered gene expression in the brain by altering BDNF activity. Similar mechanisms may underlie biomechanical processes occurring in the DRG and the dorsal horn, as BDNF and NMDA are prevalent in all neurons involved in pain transmission. We conclude that in nociceptive neurons, BDNF activity relies on DNA demethylation of the BDNF gene to prime synaptic strength and potentiation in an NMDA dependant manner.
DNA Methylation Dependant NMDA-R Variability
DNA methylation is responsible for variable NMDA receptor expression and activity. The NMDA receptor exists as a heterodimer formed by two obligatory NR1 subunits and two NR2 subunits. Different NR2 subunits confer distinct receptor binding properties. The NR2A-NR1 subtype is responsible for rapid decaying current and the NR2B-NR1 subtype for slowly decaying currents, creating an action potential that lasts more than four times longer than NR2A-NR1 generated currents. In the brain, the latter is responsible for prolonged axonal and synaptic transmission due to continuous current. Protracted potentiation is a major contributing factor to memory [62,63]. The NR2 subunits have variable binding affinities for glutamine, a known neurotransmitter in pain processing. NR2 subunits are further classified into the four subtypes: NR2A-NR2D. NR2B is the most studied subtype relevant to pain modulation. Multiple receptor isoforms with distinct central nervous system distributions and functional properties arise by differential expression of the NR2 subunit [64,65]. In experimental studies, NR2 mRNA expression in neuronal cells increased dramatically with suppression of neural activity by decreasing environmental stimulus (dark conditioning) or chemical neural blockade [64]. This effect was blunted with the addition of DNA inhibiting N-methyltransferases (DNMT), a protein responsible for methylating DNA. Interestingly, it was also found that methyl CpG binding protein 2 (MeCP2), a protein responsible for DNA silencing by DNA methylation (and secondary histone condensation and deacetylation), concomitantly bound NR2B DNA [64].
We restate the proposition that the clinical representation of LTP (Long-Term Potentiation) in nociceptive neurons may be chronic pain [66]. Therefore, we maintain that environmental factors and DNA methylation can influence NMDA directed neuronal LTP by the selective control of NMDA isoforms.
The NR2B subtype is highly expressed in the anterior cingulate gyrus (ACG), a feature that has led researchers to use it as a simplified model to study memory. The ACC is also an important structure involved in pain and emotion. Therefore, our experience of pain might be predicated, in part, to our genetically assigned variation of NMDA receptor subtypes. Thus, ACC LTP has been proposed as a cellular model to study chronic pain as well as pain-related emotional cognitive disorders.
Furthermore, environmental dictation of pain associated gene DNA methylation suggests a molecular explanation how pain plasticity can be affected by behavior and perception.
Post-Translational Modification of Histone Proteins
Histones are the principal nuclear proteins responsible for DNA condensation and decondensation. DNA is wrapped around individual histone proteins; each histone is intimately associated with pairs of four core histone proteins as well as linker and specialized variant histones. This complex arrangement of multiple histones and interlaced DNA (147 base pairs) comprises a nucleosome, the structural constituent mediating the activation status and fidelity of processing of individual transcriptional units. Each histone type possesses complementary chemical and structural properties. Histones have basic N-terminal tails that preferentially undergo multiple post-translational modification (PTM) events (acetylation, methlation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, deamination, and proline isomerization) to determine the conformational properties, environmental responsiveness, transcriptional activity, and additional epigenetic modifications of DNA. Alterations of the N-terminal tails of histones modulate the strength and flexibility of nucleosomal-DNA bonds. These examples of chromatin remodeling are directed by histonemodifying enzymes [67-69].
In transcriptionally “poised” and active chromatin regions, histone modification is a dynamic process associated with evolving steady state signatures of methylation, acetylation, phosphorylation, and associated PTMs [67,70,71].
The elegance of normal gene expression profiles and associated epigenetic regulatory mechanisms have been identified in brain and spinal cord pain centers. The deregulation of these genomic processes has been identified in disorders that relate to abnormal sensation. For example, inflammation, descending pain circuitry, and dopamine levels have underlying epigenetic mechanisms which affect pain processing [72-74]. Perhaps histone modification can explain transcription-associated gene regulation.
Behavior, Environment, and Genetic Variability
Histone PTMs can be transacted by intracellular and environmental mechanisms. RAS-MAPK, PKA, PKC, and MAPK-p38 intracellular signal transduction pathways are intimately associated with histone PTMs, thus coupling extracellular, environmentally and behaviorally derived changes to intranuclear changes which can alter the cell’s behavior and phenotype. Furthermore, histone acetylation and phosphorylation have been observed in inflammatory conditions that upregulate IL-1, IL-6, IL-8, and metalloproteinases [75]. Heat shock proteins can directly or indirectly promote the acetylation and phosphorylation of histones [76-78]. These observations suggest that stimuli damaging to the organism can directly initiate nuclear modifications by epigenetically determined mechanisms. We see that environmental changes can affect the experience of pain by modifying gene expression in a histone dependent manner. Thus, if transcription is left constituently active due to epigenetic factors, chronic pain may result. Pathologic synaptic plasticity in the brain has been associated with drug addiction, and behavioral and emotional disorders, all of which have been linked our perception of pain.
PTM of histones is an integral mechanism for facilitating neuronal development and mature homeostasis and plasticity. Dysregulation of histone PTMs has been linked to bipolar mood disorder and schizophrenia by abnormal DNA modulation and synaptic plasticity. Both psychiatric conditions have been strongly associated with atypical pain sensitivity.
Intranuclear Modifications and the NMDA Receptor
Histone PTMs of NMDA receptors can be transacted by intracellular and environmental mechanisms and have been associated with learning, memory, and behavior [79]. Specifically, H3 histone acetylation in the CNS, a clinical correlate of memory and fear conditioning, enhances LTP of CNS neurons. The enzymes responsible for NMDA coupling and histone modification have been preserved through primate evolution. This attests to their significance in human cognition and associated neuronal plasticity. Therefore, NMDA directed pain imprinting can be expanded to central nervous system pain modulation.
Our perception of acute pain may be due to rapid, short lived synaptic and axonal currents, whereas chronic pain may be electrochemically represented by prolonged signal transmission of synaptic and membrane depolarization, by slowly decaying currents. This is akin to how memory formation occurs through the mediation of slowly decaying NMDA current [2]. In fact, some studies suggest that our interpretation of acute and chronic pain may be, in part, due to the presence of varying combinations of these channel proteins [80]. Active NMDA receptors express a constant NR1 subunit with variable NR2A and NR2B subunits. NMDA receptors endowed with relatively more NR2B subunits have been linked to chronic pain [81]. In the Anterior Cingulate Cortex (ACC), a critical region of higher pain processing NMDA receptor variability has been shown to affect persistent pain by modulating axonal potentiation. NMDA associated signal transmission in the ACC has been shown to promote transcription of AMPA receptors and NR2B subtypes via CASK-interacting nucleosome assembly protein (CINAP) [82]. This phenomenon has been documented in the brain and spinal cord dorsal horn, representing a molecular model for pain potentiation via NMDA directed intranuclear modification.
NMDA directed histone modification, as it relates to pain memory and chronic pain, indicates that our pain recognition neural systems have an inherent mechanism that can alter neuronal phenotype and function by the modulation of gene function. Furthermore, environmental and behavioral processes are intimately linked to NMDA activation and can effect genetic processing. We suggest that central NMDA-receptor activation during nociception may be responsible for induction of pain memory via intranuclear aberrations.
NMDA Receptor Associated Long-term Potentiation (LTP) and Histone Modification
LTP refers to persistent enhancement of synaptic strength or depolarization following repetitive stimulation of a chemical synapse or simultaneous stimulation of two nerve cells, and may be associated with memory formation [83]. It has been reported that in pain modulating dorsal horn cells, NMDA receptoractivated signal transduction pathways facilitate activity of factors which promote LTP at nociceptive nerve fibers [84-86]. Therefore, dorsal horn cells may be responsible for “sharp pain” memory and recognition.
In the hippocampus and thalamus, NMDA receptor activation is associated with LTP and memory formation [69]. From what we know about brain related NMDA activity, we can deduce that in the spinal cord NMDA activity controls a self-amplifying and perpetuating phenomenon that is embedded within the molecular matrix of memory formation and consolidation, thus linking the molecular mechanisms of acute and chronic pain. NMDA coupling is a mechanism by which we remember from our environment if the appropriate stimulus is introduced, suggesting that we may process pain mechanistically similar to higher cerebral centers.
Central Sensitization and Histone Modification
Central sensitization refers to a continued response to pain after a painful stimulus has been removed [87,88]. Central sensitization is the consequence of ligand-receptor mediated activation of substance P/NK-1, glutamate/NMDA, and brainderived neurotrophic factor (BDNF)/tropomyosin-related kinase receptor B (TrkB) within the dorsal horn [89]. Ligand binding decreases the threshold for neural activation and the spinal cord thereafter converts an innocuous stimulus into a painful one. Activation of these receptors causes an increase in intracellular calcium [90]. As a result, there is activation of calcium-dependent kinases that phosphorylate a myriad of membrane-associated, intracellular, and nuclear proteins.
Calcium is responsible for pain mediation at extranuclear and intranuclear levels. Seconds to minutes after C-fiber stimulation there is intranuclear activation of factors that facilitate gene transcription via calcium-mediated cAMP responsive element binding protein (CREB). The transcription co-activator calcium binding proteins (CBP) in the hippocampus and laminae II of the dorsal horn was identified as a nuclear factor that binds phosphorylated CREB [91]. CBP has histone acetylating activity, which alters DNA-histone binding characteristics and directs transcription. This suggests that there is rapid and tightly coupled genetic regulation to an inciting stimulus at the level of spinal cord transduction and brain perception by a calcium mediated mechanism.
Hours after an initial painful response, calcium influx continues to phosphorylate a host of transcription factors within nociceptive neurons. The phosphorylation of these pathways has shown a direct influence on histone-associated changes in gene transcription related proteins within the DRG and dorsal horn neurons (Figure 3A-D). Brain derived neurotrophic factor (BDNF), a dynamic gatekeeper of neural plasticity, mRNA increases within two hours of C-fiber stimulation as a result of calcium influx through voltage-gated calcium channels in the DRG. BDNF is a critical mediator of memory formation in the brain and nociceptive transduction in the spinal cord. In the hippocampus, memory, learning, and fear conditioning have been linked to BDNF gene directed histone modification and NMDA receptor activity. Concordantly, the nociceptive properties of the DRG and dorsal horn have been shown to be BDNF and NMDAdependant. Keeping in mind that NMDA activation directs histone modification, which, in turn directs BDNF production we suggest that pain memory may be due to modification of histone proteins at the level of DRG, dorsal horn, and higher brain centers (Figure 3).
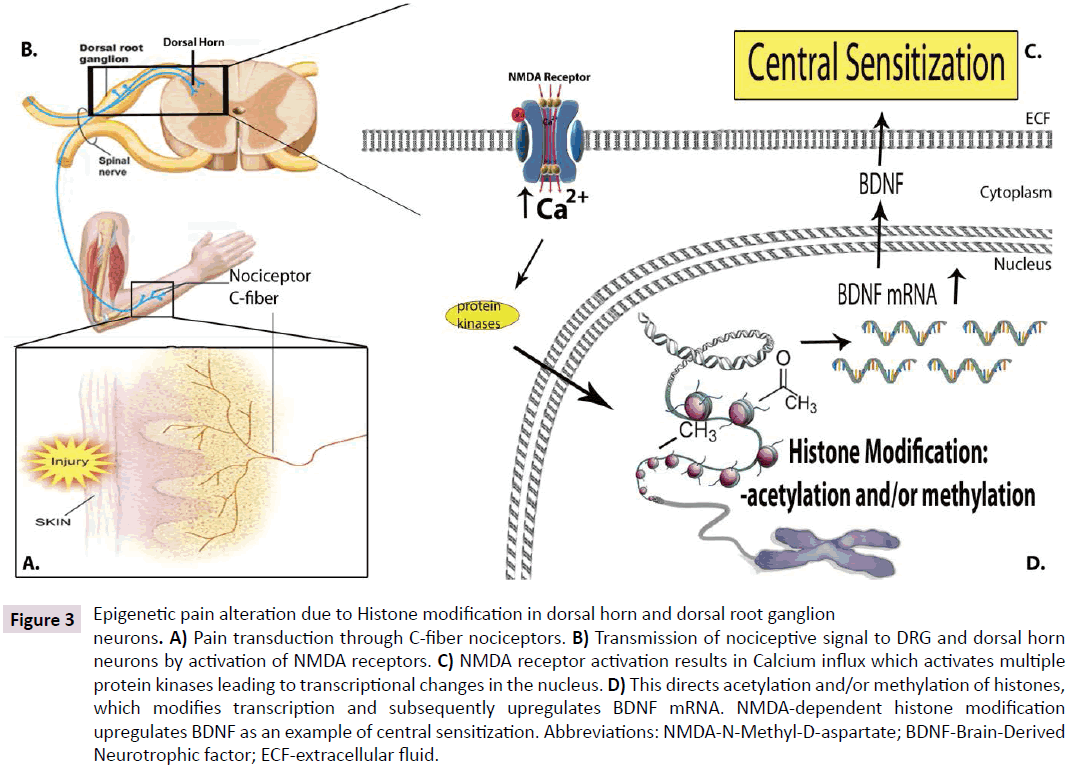
Figure 3: Epigenetic pain alteration due to Histone modification in dorsal horn and dorsal root ganglion neurons. A) Pain transduction through C-fiber nociceptors. B) Transmission of nociceptive signal to DRG and dorsal horn neurons by activation of NMDA receptors. C) NMDA receptor activation results in Calcium influx which activates multiple protein kinases leading to transcriptional changes in the nucleus. D) This directs acetylation and/or methylation of histones, which modifies transcription and subsequently upregulates BDNF mRNA. NMDA-dependent histone modification upregulates BDNF as an example of central sensitization. Abbreviations: NMDA-N-Methyl-D-aspartate; BDNF-Brain-Derived Neurotrophic factor; ECF-extracellular fluid.
This implies a novel way to consider pain transduction and perception. We have proposed that pain recognition is a DNA transcriptive process that may be modified by environmental cues and mediated by NMDA dependant histone modification of BDNF genes (Figure 3). However, histone modification can be reversed by endogenous enzymes which dissolve the histone bonds responsible for increased transcriptional activity [92,93]. Alternatively, it is reasonable to infer that the etiology of chronic pain states subsequent to an acute insult to the organism may be due to defective reversal of the histone modification pathway. The sensation of acute pain may be enhanced by NMDA receptor activity and embedding pain in memory via BDNF mediated transcription by constituently active pain modulating enzymes.
Melatonin-induced Histone Modification and Pain
Melatonin is a hormone that is primarily produced in the pineal gland, skin, eye, and gastrointestinal tract of primates. It is thought to be responsible for circadian rhythms and antioxidant properties specific to DNA [94]. Many researchers believe that melatonin mediates inflammatory and neuropathic pain modulation by interaction with melatonin receptors located in the thalamus, hypothalamus, dorsal horn, and trigeminal nucleus. It is thought that these receptors exert their anti-nociceptive influence by cAMP-mediated activation of opioid channels, activation of hyperpolarizing potassium and GABA channels, and inhibition of the pro-inflammatory mediators, 5-lipoxygenase and cyclooxygenase 2. In clinical trials, melatonin has been successfully used to treat abdominal pain in inflammatory bowel syndrome, fibromyalgia, and chronic headache [95-98].
Melatonin secretion is known to be stress and sleep dependent [99]. Pulsed melatonin is seen in the non-stressed subjects, while continuous secretion has been documented in sleep deprived individuals [100-102]. Opposing effects of continuous versus pulsed melatonin secretion suggest evidence for enduring links between pain and circadian rhythms and hormone regulation.
Recent reports indicate that melatonin is responsible for neuronal stem cell differentiation by epigenetic gene regulation. Physiological pulsed concentrations of melatonin affected epigenetic regulation by lowering the rate of histone acetylation, a primer of DNA transcription [103-108]. Melatonin administration increased mRNA expression for several histone deacetylase (HDAC) isoforms. In contrast, continuous infusion caused a significant increase in histone acetylation. Both epigenetic changes are thought to be transacted by melatonin receptors. Therefore, melatonin induced pain suppression may be mediated, in part, by the epigenetic modulation of GABAergic, opiate, and inflammatory receptor gene transcription. Conceivably, this may be via a melatonin mediated, histone acetylation pathway which relies on an environmental cue such as sleep to modify pain perception.
Thus, we propose that behavior and environmental patterns may affect DNA processing through histone modifications. This effect alters interneuronal communication and intraneuronal information transfer, altering pain processing. Accordingly, aberrations of this process can potentiate and sustain pain, producing chronic pain states.
Non-coding RNA
Intronic sequences of RNA, once thought to be inconsequential elements of the DNA sequence within protein encoding loci, are now believed to be implicated in differential modulation of the DNA transcript. Intronic sequences have been preserved in the mammalian genetic code, thereby attesting to their importance in the role of human development. These RNA sequences are called non-coding RNAs (ncRNAs). ncRNAs are variable length RNA which are not translated into a protein products but rather transact transcriptional, pre-translational, and translational material to promote genetic and cellular diversity. They can modify transcripted or translated DNA sequences, presumably to promote cellular and synaptic diversity. Some ncRNAs can be directly modified to mRNA, creating a new platform for protein assembly while rapidly promoting cellular plasticity [108]. In the human nervous system, they may be responsible for finetuning synaptic strength and other elements of transmission [109,110]. Examples of ncRNAs include transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), small RNAs such as small nucleolar RNAs (snoRNAs), microRNAs (miRNAs), small interfering RNAs (siRNAs), piwi protein-interacting RNAs (piRNAs), and longer ncRNAs such as Xist, Evf, and Air.
ncRNAs can lay dormant within an RNA product until an environmental cue initiates ncRNA activity. The ncRNA can then affect protein transcription or translation, in some cases producing an enzyme isotype which will alter synaptic function. In this way, ncRNAs may influence the synaptic plasticity of pain regulation [110-115]. The genetic variability of mRNA endowed with non coding RNA nucleotide segments has been demonstrated within the intronic sequences of several pain modulatory proteins including catechol-O-methyl transferase (COMT), opiate receptor genes, cannabinoid related fatty acid amino hydrolase genes (FAAH), GTP cyclohydrolase 1 genes, and inflammatory cytokine genes such as IL-1. Thus, the environment can affect RNA coding sequence and, thus, modify associated neuronal electrophysiologic and biochemical properties of pain transduction, transmission, and perception.
miRNA
miRNAs are critical components involved in inflammatory processing. microRNAs (miRNAs) are short (20–30 nucleotide) regulatory molecules that decrease the translational efficacy and the transcriptional stability of target RNAs by binding to target mRNAs (Figure 4A and B) [1076] Moreover, the mRNAs of inflammatory proteins are known to be particularly vulnerable to miRNA modification [107]. Levels of miRNA rapidly increase in sensory ganglion after induced muscle inflammation. They play an important role in pain processing and are differentially expressed in sensory ganglions depending upon the intensity of the pain stimulus [111].
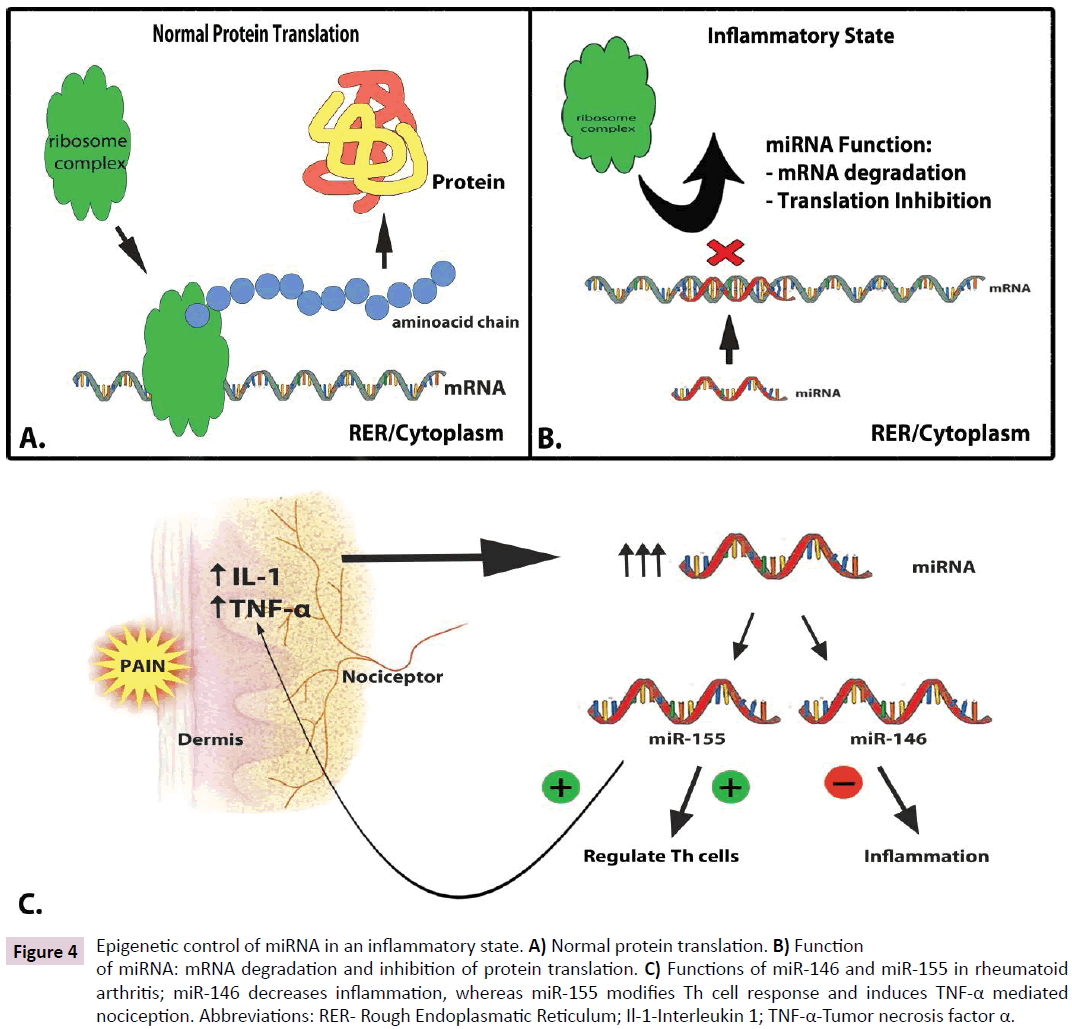
Figure 4: Epigenetic control of miRNA in an inflammatory state. A) Normal protein translation. B) Function of miRNA: mRNA degradation and inhibition of protein translation. C) Functions of miR-146 and miR-155 in rheumatoid arthritis; miR-146 decreases inflammation, whereas miR-155 modifies Th cell response and induces TNF-α mediated nociception. Abbreviations: RER- Rough Endoplasmatic Reticulum; Il-1-Interleukin 1; TNF-α-Tumor necrosis factor α.
In hematopoietic cells, miRNA levels dynamically control lineage differentiation of CD4/CD8 cells, B cells, and platelets, thus modulating inflammation at the cellular level. Abnormal expression of miRNAs has also been observed in patients with rheumatoid arthritis [112,113]. In addition, miR-146 and miR-155 are upregulated by the pain mediating cytokines IL-1 and TNFalpha (Figure 4C). A variety of miRNAs has also been identified in synovial fibroblasts and rheumatoid synovial tissue. miR-146 is responsible for inhibiting pathogen initiated inflammation. miR-155 regulates helper T cells and enhances inflammation by upregulating TNF-alpha mRNA (Figure 4C). Furthermore, spinal glial cells, recently receiving much attention for their roles in pain modulation, share many functional similarities with immune cells, including TNF-alpha and IL-6 production in response to noxious stimuli. To date, 43 types of glial miRNA have been identified. In inflammatory cells, TNF-alpha and IL-6 direct the transcription of other inflammatory pain mediators [53]. However, there is mounting evidence that these cells also react to their environment in a miRNA dependant manner. A pro-inflammatory mediator of lymphocytes, mi29a, has recently been identified as a regulatory element in myelin production and painful peripheral neuropathies [114].
Inflammatory pain transmission represents a type of homeostatic challenge. Nuclear processes and axonal transport activities are upregulated within seconds to prepare the nerve by adapting and reacting to waves of incoming nociceptive information. After injection of a known synthetic pro-inflammatory and pain evoking agent to rat facial muscles, miRNA levels were decreased within four hours with excessive rebound miRNA upregulation after four days; this expression profile inversely correlated to local inflammation. The initial miRNA downregulation stabilized inflammatory protein mRNA, allowing increased protein production. Subsequently, the rise of miRNA destabilized inflammatory mRNA and dampened the inflammatory response by inhibiting further production of inflammatory mediators.
miRNA can selectively dull or exacerbate an inflammatory response in response to noxious stimuli and has profound implications on the study of pain epigenetics. The elucidation of inflammatory mediator responsive miRNAs can potentially lead to new developments in aberrant pain and inflammatory diseases. These developments also suggest that adaptive and innate immunity are coupled at the level of mRNA translation. Therefore, miRNA deregulation may produce genomic changes that maintain inflammatory initiated pain states.
Small Nucleolar RNAs
snoRNAs are a class of small RNA molecules which regulate the modification of other RNAs and have been implicated in an array of homeostatic cellular processes including alternate RNA splicing, transcription, chromosome maintenance and segregation, genomic imprinting, and cell cycle regulation. snoRNAs display high levels of expression in the nervous system, especially in areas associated with learning and memory such as the hippocampus and cortical brain structures, suggesting that snoRNAs may be important in NMDA receptor-mediated, neuronal plasticity processes. However, snoRNA has recently been identified in pain modulating centers. snoRNA MBI36 has been identified in the thalamus, U3snoRNA in peripheral nerves, and MBII-85 in dorsal root ganglion. The discovery of snoRNAs in these sites suggests that snoRNAs may convert salient nociceptive stimuli into aversive memory traces, producing pain memory, clinically described as chronic pain. Prader-Willi Syndrome, a genetic disease characterized by hyperphagia, learning disabilities, and decreased pain sensation, is due to the absence of multiple snoRNAs in chromosome 15q11-2. Therefore, snoRNAs may be important for translating environmental cues into memory traces via NMDA receptor associated LTP.
RNA Editing
RNA editing is a dynamic process of base recoding that relies on RNA editing enzymes that modify the nucleotide sequence and therefore the functional properties and levels of expression of a spectrum of protein-coding mRNAs [115]. This epigenetic mechanism also influences the biophysical properties and functional profiles of a potentially broad array of ncRNAs and is particularly active in neurons. RNA editing in the nervous system is involved in neural development, mature steady state functions, and synaptic and neural network plasticity. RNA editing is also intimately involved in axonal myelination, thermoregulation, synaptogenesis, neural network integration, cellular stress, and energy metabolism. These are all cellular processes also associated with pain regulation.
Homer I, Homer Ia, and RNA Editing
Homer Ia is a constitutively expressed scaffolding protein found in dorsal horn neurons and promotes pain signaling [116]. It binds metabotropic glutamate receptors at the plasma cell membrane and couples signal transduction by forming a signaling complex that generates IP3 and releases calcium from intracellular pools (Figure 5A). The native Homer gene transcription product is known to undergo editing by site specific deamination to produce Homer Ia, a Homer variant with different cellular functions [117-119] Homer Ia is a dynamically regulated splice variant of Homer I expressed during states of dorsal horn hyperactivity and lacks the ability to link to glutamate receptors, dampening acute pain transmission (Figure 5B and C) [120]. Homer Ia is rapidly induced in spinal neurons after peripheral injury in an NMDA receptordependent manner [121]. Preventing upregulation of Homer Ia in mice using short hairpin RNA (shRNA) in mice exacerbates pain, and heterotopic expression of Homer Ia in specific spinal segments reduces inflammatory hyperalgesia. We see that mRNA transcript editing can affect synaptic pain transmission by protein activity modulation.
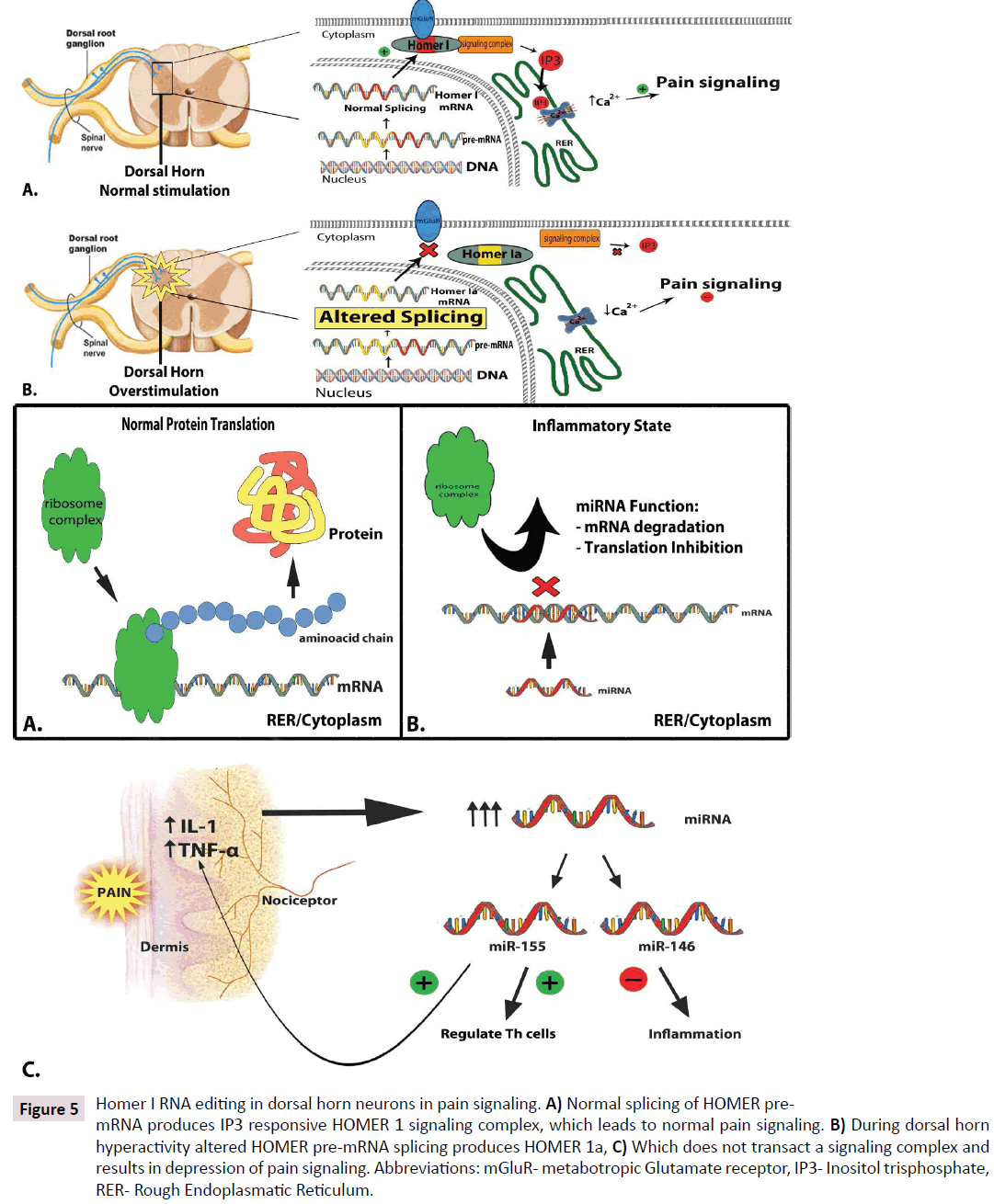
Figure 5: Homer I RNA editing in dorsal horn neurons in pain signaling. A) Normal splicing of HOMER premRNA produces IP3 responsive HOMER 1 signaling complex, which leads to normal pain signaling. B) During dorsal horn hyperactivity altered HOMER pre-mRNA splicing produces HOMER 1a, C) Which does not transact a signaling complex and results in depression of pain signaling. Abbreviations: mGluR- metabotropic Glutamate receptor, IP3- Inositol trisphosphate, RER- Rough Endoplasmatic Reticulum.
Homer Ia also strongly attenuates MAP kinase activation induced by glutamate receptors and reduces synaptic contacts on spinal cord neurons which process pain inputs. MAP kinase is an intermediate activator of gene transcription and therefore these observations suggest that RNA editing by the selective splicing of Homer I to Homer Ia mRNA can also affect pain plasticity by modulating downstream activation of pain promoting factors.
The evolutionary significance of acute pain inhibition in response to damaging stimuli allows an organism to react to its environment without the constraints of pain to hamper its decision-making. For example, pain can affect an organism’s decision to fight or flee from a situation. Moreover, the Homer gene displays very similar sequencing characteristics across vertebrates [122,123]. This suggests that Homer control has likely been preserved throughout evolution and attests to the importance of RNA editing pain modulation for survival.
RNA Editing of 5-HT
The predominant form of RNA editing in humans is adenosine to inosine deamination. In this form of RNA editing, adenosine deaminases acting on RNAs (ADARs) transact the transformation of adenosine bases into inosine bases. The base switch alters RNA translation and also modifies the final protein product, ultimately affecting cellular function. ADARs have been known to play a modulatory role in ion channels and ligand gated receptors. It has been known for some time that transcript encoding proteins involved in fast neurotransmission, including voltage-gated ion channels and neurotransmitter receptors, are subject to this type of RNA editing.
The 5-HT2C receptor is a G-protein coupled protein widely distributed throughout the spinal cord and mediates pain fibers which descend from cranial and upper spinal cord tracts. Descending pain fibers are generally known to decrease pain sensation at the level of the dorsal horn by depressing the activity of ascending, pain provoking fibers. Therefore, dysfunction of these descending regulatory tracts can cause hypersensitivity to pain. The 5-HT2C mRNA is a substrate for ADAR at five different sites. The editing sites are denoted by the letters A, B, C, D, and E. Some or all of these sites can be edited to produce up to twenty different 5-HT2C isotypes, depending upon the editing location [10]. The transcripts differ in their ability to co-activate associated G-proteins and results in increased or decreased synaptic activity. Site editing has been shown to correlate with different clinical manifestations. For example, in post mortem samples from suicide victims’ sites C and E of the mRNA transcript displayed considerable RNA editing. In a neuropathic pain model 5-HT2C mRNA displayed a reduced editing pattern at site B, producing a 5-HT2C receptor model which bound 5-HT more effectively, presumably by blocking ascending nociceptive transmission. Thus, selective ADAR editing of RNA transcripts can promote pain plasticity by altering synaptic transmission via variable 5-HT2C receptor production.
Furthermore, there is evidence to suggest that ADAR RNA editing is a responsive process to environmental cues. The catalytic core of an ADAR subtype, ADAR2, is inositol hexakisphosphate, a molecule which harvests phosphate ions for immediate cellular energy utilization, enzyme activation, and cell signaling [124-126]. The inositol hexakisphosphate moiety endows ADAR2 with the ability to expeditiously react to incoming stimuli by the selective modification of RNA and subsequent generation of variable synaptic proteins to produce cellular responses. This type of emergent synaptic plasticity is a required for an organism to react to dynamic environmental changes. RNA modification by ADAR is an environmentally responsive element which directs, in part, pain plasticity. Moreover, it is possible that ADARs activity is genetically determined, individually variable, and stimuli specific, thus providing an explanation why similar painful stimuli can provoke varying responses in different subjects.
RNA Editing and Spinal Glia
There is a rapidly growing body of evidence indicating that microglia play an important role in pain modulation. Glial cells are non-neuronal cells of the central nervous system and perform a myriad of neuronal regulatory and maintenance functions. They are responsible for neighboring axonal and dendritic growth, nutrient delivery, synaptic neurotransmitter uptake, and neuronal migration early in development. Supporting glial cells, their receptors, and secreted mediators release nitric oxide, excitatory amino acids, and pro-inflammatory cytokines that influence pain [127-132]. The cells have also been shown to act as key contributors to acute and chronic cellular pain transduction in spinal cord and brain centers due to their function as neuronal modulators [133]. Thus, drugs that block glial cell activation and the release of neuromodulators could have therapeutic potential in acute and chronic pain treatment.
The ability of glial cells to relay synaptogenic and axonal information to surrounding elements gives glia a characteristically neuronal character. However, glial cells are histologically and functionally similar to immune cells. They are scavengers of the nervous system that must be able to recognize and terminate an array of offending pathogens and damaged nerve cells. Furthermore, glia perform cell signaling in a manner similar to their immune cells counterparts, which constantly vary their cellular markers to communicate with other cells [134-139].
There are similarities in glial and immune cell behavior which suggests that they may share molecular characteristics which generate plasticity. It has been postulated that in antibody producing B cells, edited RNA sequences are written back to DNA to induce phenotypic and morphological changes that alter DNA sequences for the purpose of generating receptor diversity. These changes are considered heritable and evolutionarily viable because they have been inscribed into the genetic material. Permanence of the genetic code is dictated by subsequent mRNA DNA encoding by native reverse transcriptases or via RNA-directed DNA repair pathways. Both processes have been described to occur in immune and nerve cells and are transacted by the apolipoprotein B mRNA editing enzyme catalytic polypeptides (APOBEC) class of cytidine deaminases that perform C-U C-T editing of mRNA and subsequent re-writing of the genetic code by DNA polymerases. APOBEC enzymes show some of the strongest signatures of positive selection in human immune systems suggesting that they are designed for adjusting to changing environmental and pathogenic states [140,142]. Thus, the action of APOBEC on mRNA transcription are thought to support adaptive changes in human neurons and immune cells [143-146]. In fact, APOBEC deregulation does occur and has been associated with tumor growth and neuropathic pain [147-150].
We present the idea that glia are dynamic modulatory cells constantly changing in response to environmental stimuli due to mRNA editing and that these altered RNA transcripts can be written back to the DNA for permanent expression. The ramifications of such a supposition are important towards our understanding of and treatment of chronic pain [151]. It suggests that a defect in mRNA editing or DNA encoding in glial cells can promote deregulated pain. Thus, chronic pain may be due to pathological RNA editing and DNA rewriting, which affects glial cell activity by permanently modifying the genome.
Synaptic RNA and Long-term Facilitation
Though once believed that RNA was exclusively a nuclear substrate, recent experimental evidence suggests otherwise. Numerous ncRNAs have been identified in the cytoplasm, axons, and peri-synaptic clefts of glial neurons. Nuclear-cytoplasmic (axodendritic) translocation occurs under homeostatic conditions in preparation for local synaptic-associated stimuli. Based on empirical evidence and theoretical grounds, it is generally agreed that RNA editing occurs in these areas to modulate synaptic activity and connectivity in response to local environmental inputs. The teleological significance of peridendritic RNA might be for the promotion of rapid synaptic plasticity to accommodate an organism’s neurological response to potentially threatening external stimuli. These synaptic changes might mediate memory by re-writing synaptic changes into the cell’s genetic architecture. Accordingly, chronic pain may, in part, be due to RNA associated synaptic translation executed through synaptic RNA directed receptor integration, disintegration, or receptor modulation. Data demonstrate that CNS axons contain many mRNA species of diverse functions, and suggest that, like invertebrate and PNS axons, CNS axons synthesize proteins locally, maintaining a degree of autonomy from the cell body [152,153].
Short-term synaptic changes (STSC) are molecularly distinguished from long- term synaptic changes (LTSC). The former process modifies existing synaptic proteins, whereas the latter activates synaptic protein synthesis. In a study to elucidate peridendritic protein synthesis in synaptic plasticity, LTSC was linked to local synaptic protein transcription and translation [154]. Synaptic transmission in hippocampal cells were increased when the neurotrophic factors BDNF and NT-3 were introduced to neurons with chemically or physically lesioned cell bodies to disable nuclear directed protein assembly. Subsequent addition of protein translation inhibitors did not affect protein modification but blocked long-term synaptic changes. This evidence not only shows that elements of protein assembling machinery exist outside of the nucleus, such as RNA derived protein assembly elements tRNA and polyribosomes, but that BDNF and NT-3 may regulate local synaptic plasticity. Recently, an investigation of synaptic translation and localization of synaptic LTSC mRNAs displayed that peridendritic protein synthesis was mediated by BDNF, NMDA, and serotonin in a spatially restricted, stimulus specific manner [154]. Sensorin mRNA, the precursor of a sensory nerve specific neurotransmitter, localized to distal neuronal processes and was shown to have variable synaptic destinations predicated by the mRNA sequence. 5’UTR capping of mRNA directed sensorin mRNA to synapses while 3’UTR capping localized the mRNA to neurites. Subsequent site specific synaptic serotonin perfusion of neurons (without a cell body) increased sensorin translation in associated areas indicating that serotonin mediated localized synaptic protein processing. Thus, we see that differential RNA splicing might be responsible for targeting mRNA to designated sites for processing and that serotonin mediated the conversion from precursor elements to protein, thereby affecting neuronal plasticity. Furthermore, the injection of calcium binding agents in post-synaptic nerves completely inhibited serotonin induced pre-synaptic translation of sensorin without affecting perisynaptic localization, thereby suggesting that retrograde calcium signaling is a necessary component of RNA directed synaptic translation.
BDNF, NT-3, and serotonin are chemical initiators of synaptic signaling and mediators of pain processing. Thus, we suggest that the aforementioned pathways are also involved in pain transmission. Most of the studies displaying synaptic translation have been done in brain regions associated with memory, suggesting a role for long-term pain potentiation and pain perception. Further investigation is needed to investigate whether spinal cord elements may also be involved.
The relevance of synaptic RNA in nociceptive fibers has yet to be studied; however, our growing understanding of the glial cell role in pain transmission and their newfound discovery of peri-synaptic RNA activity suggests that this phenomenon may occur in pain centers. It is conceivable that peri-dendritic glial protein synthesis directly or indirectly affects neighboring axons’ morphological and electrochemical properties by local neurotrophic factor production and receptor modification. Furthermore, it suggests that glial cells may contribute to nociception in a way not previously described. The implication of synaptic RNA directed protein integration and assembly for the nociceptive system is remarkable, as it describes a new mechanism to explain acute and chronic pain.
The role of pain RNA mediated synaptic facilitation may best be exemplified by examining the role of glia in pain transmission. Earlier we stated that glia have dendritic extensions that communicate with axons and synapses of upper motor neurons. They have been shown to modulate pain by serotonergic inhibitory pathways. Therefore, a disruption of this pathway may lead to pain hypersensitivity. Glia cells affect pain processing both pre-synaptically by modifying ligand production and postsynaptically by changing the expression of the corresponding receptors as well as displaying high levels of perisynaptic and axonal activity during pain transmission. Furthermore, RNA and serotonin have been identified in their distal dendrites and axons, suggesting that the extra axonal translational processes aforementioned might be responsible for the maintenance of a painful stimulus by molecular changes that permanently alter the synaptic interface. Thus, we propose that glia may affect their neuronal networks via epigenetic interaction by effector RNA, producing translational changes that modify their synapses to promote LTSC. In the context of pain signaling, LTSC describes constant depolarization of pain signals, thereby suggesting a clinical correlate of chronic pain. In many chronic pain states such as Chronic Regional Pain Syndrome (CRPS), an acute noiceptive event often precedes maintenance (chronic) pain. Associated hyperalgesia and allodynia soon follows, however, in a time frame too short to be explained by the classical model of synaptic plasticity by nuclear transcription and cytoplasmic translation. Thus, we propose that specialized brain and spinal cord centers promote peri-synaptic, RNA directed rapid synaptic change to maintain and propagate a noxious stimulus. It follows that a defect in this process can propagate LTSC of pain, causing clinically identifiable chronic pain.
Further study of dendritic RNA mechanisms and dynamics may help to elucidate new therapies for hyperalgesia [14].
Conclusion
Whether we are discussing the peripheral axon, dorsal root ganglion, dorsal horn, midbrain, pons, or supratentorial cells, there is no doubt that pain neurons are unusually metabolically active particularly when stimulated. Nociceptive cells constantly receive peripheral cues that are relayed to the nucleus; the nucleus then relays the cell signal to the next higher-order neuron within an integrated neural network. In most cases, information travels a considerable distance within peripheral nerve and spinal cord tracts before nuclear changes occur. Activation of different nociceptors produces varying genomic alterations. For example, we have seen that inflammatory molecules may produce phenotypic changes in the form of receptor modifications, while chemical and thermal stimuli produce immediate axonal changes that clinically manifest as acute pain. However, constant activation of pain receptors can subsequently lead to long-term activation of nuclear mediators that may induce chronic and hyperactive pain syndromes. Nociceptive nerves, similar to cerebral nerves, have the capacity to learn and change phenotypes. The dysregulation of these integrated processes may lead to chronic pain.
The epigenetic control of the nociceptive system is an elegant model to describe how the environment and behavior affects pain perception. Plasticity and memory formation of the pain neuronal network is executed by changes on a RNA and chromatin level and subsequently modulates the genome.
Whether a pain response is immediate or prolonged, there is evidence for epigenetic modulation. ncRNAs are present within synapses, dendrites, axons, and perinuclear and intranuclear spaces. This attests to the extraordinary range and versatility of RNA functions and the wide array of RNA dependent intraand inter-cellular processes. RNA can affect pain perception by modulating ion channels and transcription factors, transport signaling molecules, and edited pre-translated and non-coding RNAs to produce pain receptor proteins and RNA species with different degrees of activity. This directly modulates pain-related protein and ncRNA production in a nucleosomal-dependent manner, and may even act as dynamic ligands for mediating pain processing.
The epigenetic model of pain signaling provides a new, plausible, and elegant model to describe the relationship between acute and chronic pain. Furthermore, the disruption of these intracellular epigenetic networks may explain individual pain perception and diseases associated with persistent hyperalgesic states. Perhaps the key to treating chronic pain lies in our understanding of epigenetics and its effects on cellular function.
Acknowledgements
There was no conflict of interest in the production of this review.
References
- Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D (2006) Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain 10: 287-333.
- Tran L, Chaloner A, Sawalha AH (2012) Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuromodulation 38: 898-906.
- Woolf CJ, Costigan M (1999) Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci U S A 96: 7723-7730.
- Bromage PR (1973) Nerve physiology and control of pain. Orthop Clin North Am 4: 897-906.
- Diatchenko L, Nackley AG, Tchivileva IE, Shabalina S A, Maixner W (2007) Genetic architecture of human pain perception. Trends Genet 23: 605-613.
- Dwarakanath G K, Warfield C A (1986) The pathophysiology of acute pain. Hosp Pract (Off Ed) 21: 64B, 64G-64H, 64L passim.
- Sittl R, Boujong D, Griessinger N (1996) Physio- and pathophysiology of acute pain with special consideration of pain prevention. Anaesthesist, 45 Suppl 3: S72-73.
- Woolf C J (1989) Recent advances in the pathophysiology of acute pain. Br J Anaesth 63: 139-146.
- Bruehl S, Chung O Y, Burns JW (2003) Differential effects of expressive anger regulation on chronic pain intensity in CRPS and non-CRPS limb pain patients. Pain, 104: 647-654.
- Frettloh J, Huppe M, Maier C (2006) Severity and specificity of neglect-like symptoms in patients with complex regional pain syndrome (CRPS) compared to chronic limb pain of other origins. Pain, 124: 184-189.
- Geha PY, Baliki MN, Harden RN, Bauer WR, Parrish TB et al. (2008) The brain in chronic CRPS pain: abnormal gray-white matter interactions in emotional and autonomic regions. Neuron, 60: 570-581.
- Lipov E G, Joshi JR, SandersS, Slavin KV (2009) A unifying theory linking the prolonged efficacy of the stellate ganglion block for the treatment of chronic regional pain syndrome (CRPS), hot flashes, and posttraumatic stress disorder (PTSD). Med Hypotheses.
- Buskila D (2007) Genetics of chronic pain states. Best Pract Res Clin Rheumatol, 21: 535-547.
- Descalzi G, Ikegami D, Ushijima T (2015) Epigenetic mechanisms of chronic pain. Trends Neurosci, 38: 579.
- Stephan M, Helfritz F, Pabst R, von Horsten S (2002) Postnatally induced differences in adult pain sensitivity depend on genetics, gender and specific experiences: reversal of maternal deprivation effects by additional postnatal tactile stimulation or chronic imipramine treatment. Behav Brain Res, 133: 149-158.
- Mattick JS, Mehler MF (2008) RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci, 31: 227-233.
- Esteller M (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet, 8: 286-298.
- Montagna P (2008) The primary headaches: genetics, epigenetics and a behavioural genetic model. J Headache Pain, 9: 57-69.
- Ceyhan GO, Deucker S, Demir IE, Erkan M, Schmelz M, et al. (2009) Neural fractalkine expression is closely linked to pain and pancreatic neuritis in human chronic pancreatitis. Lab Invest, 89: 347-361.
- Ceyhan GO, Michalski CW, Demir IE, Muller MW, Friess H (2008) Pancreatic pain. Best Pract Res Clin Gastroenterol, 22: 31-44.
- Elliott MB, Barr AE, Clark BD, Amin M, Amin S, et al. (2009) High force reaching task induces widespread inflammation, increased spinal cord neurochemicals and neuropathic pain. Neuroscience, 158: 922-931.
- Waldmann R, Lazdunski M (1998) H(+)-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol, 8: 418-424.
- Walker K, Perkins M, Dray A (1995) Kinins and kinin receptors in the nervous system. Neurochem Int, 26: 1-16; discussion 17-26.
- Wilson AG (2008) Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J Periodontol, 79: 1514-1519.
- Dray A (1995) Inflammatory mediators of pain. Br J Anaesth, 75: 125-131.
- Tate SS, Sweet R, McDowell FH, Meister A (1971) Decrease of the 3,4-dihydroxyphenylalanine (DOPA) decarboxylase activities in human erythrocytes and mouse tissues after administration of DOPA. Proc Natl Acad Sci U S A, 68: 2121-2123.
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, et al. (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron, 21: 531-543.
- Koltzenburg M, Lundberg LE, Torebjork HE (1992) Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain, 51: 207-219.
- Stubhaug A (1997) A new method to evaluate central sensitization to pain following surgery. Effect of ketamine. Acta Anaesthesiol Scand Suppl, 110: 154-155.
- Stubhaug A, Breivik H (1997) Long-term treatment of chronic neuropathic pain with the NMDA (N-methyl-D-aspartate) receptor antagonist ketamine. Acta Anaesthesiol Scand, 41: 329-331.
- Mantyh PW, Koltzenburg M, Mendell LM, Tive L, Shelton DL. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. (2011). Anesthesiology. 115: 189-204.
- Davis RJ (1993) The mitogen-activated protein kinase signal transduction pathway. J Biol Chem, 268: 14553-14556.
- Abdolmaleky HM, Smith CL, Zhou JR, Thiagalingam S (2008) Epigenetic alterations of the dopaminergic system in major psychiatric disorders. Methods Mol Biol 448: 187-212.
- Pineyro G, Archer-Lahlou E (2007) Ligand-specific receptor states: implications for opiate receptor signalling and regulation. Cell Signal, 19: 8-19.
- Ambriz-Tututi M, Rocha-Gonzalez HI, Cruz SL, Granados-Soto V (2009) Melatonin: A hormone that modulates pain. Life Sci 84: 489-498.
- Formisano L, Noh KM, Miyawaki T, Mashiko T, Bennett M V, et al. (2007) Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampal neurons. Proc Natl Acad Sci U S A, 104: 4170-4175.
- Li-Na (2008) Epigenetic control of the expression opioid receptor genes epigenetics, 3: 119-121.
- Olmstead MC, Ouagazzal AM, Kieffer BL (2009) Mu and delta opioid receptors oppositely regulate motor impulsivity in the signaled nose poke task. PLoS ONE, 4: e4410.
- Lal G, Zhang N, van der Touw W, Ding Y, Ju W, et al. (2009) Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol, 182: 259-273.
- Colafrancesco V, Cirulli F, Rossi S, Berry A, Aloe L (2009) Anti-NGF-antibody administration as collyrium reduces the presence of NGF and enhances the expression of VEGF in the retina, lacrimal gland and hippocampus. Neurosci Lett, 463: 203-206.
- Cunha C, Brambilla R, Thomas KL (2010) A simple role for BDNF in learning and memory? Front Mol Neurosci, 3: 1.
- Valdes-Sanchez T, Kirstein M, Perez-Villalba A, Vega JA, et al. (2010) BDNF is essentially required for the early postnatal survival of nociceptors. Dev Biol.
- Ognibene E, Adriani W, Granstrem O, Pieretti S, Laviola G (2007) Impulsivity-anxiety-related behavior and profiles of morphine-induced analgesia in heterozygous reeler mice. Brain Res, 1131: 173-180.
- Akopians AL, Babayan AH, Beffert U, Herz J, Basbaum AI, et al. (2008) Contribution of the Reelin signaling pathways to nociceptive processing. Eur J Neurosci 27: 523-537.
- Villeda SA, Akopians AL, Babayan AH, Basbaum AI, Phelps PE (2006) Absence of Reelin results in altered nociception and aberrant neuronal positioning in the dorsal spinal cord. Neuroscience, 139: 1385-1396.
- Gerber G, Youn DH, Hsu CH, Isaev D, Randic M (2000) Spinal dorsal horn synaptic plasticity: involvement of group I metabotropic glutamate receptors. Prog Brain Res, 129: 115-134.
- Ikeda H, Heinke B, Ruscheweyh R, Sandkuhler J (2003) Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science, 299: 1237-1240.
- Ikeda H, Kiritoshi T, Murase K (2009) Synaptic plasticity in the spinal dorsal horn. Neurosci Res, 64: 133-136.
- Ma JY, ZhaoZQ (2002) The involvement of glia in long-term plasticity in the spinal dorsal horn of the rat. Neuroreport, 13: 1781-1784.
- Moore KA, Baba H, Woolf CJ (2000) Synaptic transmission and plasticity in the superficial dorsal horn. Prog Brain Res 129: 63-80.
- Zhang HM, Qi YJ, Xiang XY, Zhang T, Liu XG (2001) Time-dependent plasticity of synaptic transmission produced by long-term potentiation of C-fiber evoked field potentials in rat spinal dorsal horn. Neurosci Lett 315: 81-84.
- Cervenka S, Palhagen SE, Comley RA, Panagiotidis G, Cselenyi Z, et al. (2006) Support for dopaminergi hypoactivity in restless legs syndrome: a PET study on D2-receptor binding. Brain 129: 2017-2028.
- Flores JA, El Banoua, Galan-Rodriguez B, Fernandez-Espejo E (2004) Opiate anti-nociception is attenuated following lesion of large dopamine neurons of the periaqueductal grey: critical role for D1 (not D2) dopamine receptors. Pain 110: 205-214.
- Potvin S, Grignon S, Marchand S (2009) Human evidence of a supra-spinal modulating role of dopamine on pain perception. Synapse 63: 390-402.
- Wood PB, Schweinhardt P, Jaeger E, Dagher A, Hakyemez H, et al. (2007) Fibromyalgia patients show an abnormal dopamine response to pain. Eur J Neurosci 25: 3576-3582.
- Burkey AR, Carstens E, Jasmin L (1999) Dopamine reuptake inhibition in the rostral agranular insular cortex produces antinociception. J Neurosci 19: 4169-4179.
- Buskila D (2007) Genetics of chronic pain states. Best Pract Res Clin Rheumatol 21: 535-547
- Shyu BC, Kiritsy-Roy JA, Morrow TJ, Casey KL (1992) Neurophysiological, pharmacological and behavioral evidence for medial thalamic mediation of cocaine-induced dopaminergic analgesia. Brain Res 572: 216-223.
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, et al. (2008) Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 82: 696-711.
- Veldic M, Kadriu B, Maloku E, Agis-Balboa RC, Guidotti A, et al. (2007) Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr Res 91: 51-61.
- Lubin FD, Roth TL, Sweatt JD (2008) Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 28: 10576-10586.
- Lisman JE, Fellous JM, Wang XJ (1998) A role for NMDA-receptor channels in working memory. Nat Neurosci 1: 273-275.
- Yamada k, Mizuno M, Nabeshima T (2002) Role for brain-derived neurotrophic factor in learning and memory. Life Sci 70: 735-744.
- Lee S, Kim W, Ham BJ, Chen W, Bear MF, et al.(2008) Activity-dependent NR2B expression is mediated by MeCP2-dependent epigenetic regulation. Biochem Biophys Res Commun 377: 930-934.
- Nakae A, Nakai K, Tanaka T, Takashina M, Hagihira S, et al. (2008) Serotonin2C receptor mRNA editing in neuropathic pain model. Neurosci Res 60: 228-231.
- Taniura H, Sng JC, Yoneda Y (2007) Histone modifications in the brain. Neurochem Int 51: 85-91.
- Chung D (2002) Histone modification: the 'next wave' in cancer therapeutics. Trends Mol Med 8: S10-11.
- Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, et al. (2008) The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol 15: 1968-1976.
- Seligson DB, Horvath S, Shi T, Yu H, Tze S, et al. (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435: 1262-1266.
- Kristeleit R, Stimson L, Workman P, Aherne W (2004) Histone modification enzymes: novel targets for cancer drugs. Expert Opin Emerg Drugs 9: 135-154.
- Majid S, Dar A A, Ahmad A, Hirata H, Kawakami K, et al. (2009) BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 30: 662-670.
- Martinowich K, Hattori D, Wu H, Fous S, He F, et al. (2003) DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302: 890-893.
- Nakae A, Nakai K, Tanaka T, Hagihira S, Shibata M, et al. (2008) The role of RNA editing of the serotonin 2C receptor in a rat model of oro-facial neuropathic pain. Eur J Neurosci 27: 2373-2379.
- Rygh LJ, Suzuki R, Rahman W, Wong Y, Vonsy JL, et al. (2006) Local and descending circuits regulate long-term potentiation and zif268 expression in spinal neurons. Eur J Neurosci 24: 761-772.
- Bi Y, Liu G, Yang R (2009) MicroRNAs: novel regulators during the immune response. J Cell Physiol 218: 467-472.
- Birnie DH, Vickers LE, Hillis WS, Norrie J, Cobbe SM (2005) Increased titres of anti-human heat shock protein 60 predict an adverse one year prognosis in patients with acute cardiac chest pain. Heart 91: 1148-1153.
- Khan IU, Wallin R, Gupta RS, Kammer GM (1998) vProtein kinase A-catalyzed hosphorylation of heat shock protein 60
- chaperone regulates its attachment to histone 2B in the T lymphocyte plasma membrane. Proc Natl Acad Sci USA 95: 10425-10430.
- Shams S, Shafi S, Bodman-Smith K, Williams P, Mehta S, et al. (2008). Anti-heat shock protein-27 (Hsp-27) antibody levels in patients with chest pain: association with established cardiovascular risk factors. Clin Chim Acta 395: 42-46.
- Chen L, Huang L Y (1992). Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature 356: 521-523.
- Willis WD, Jr (1986) Visceral inputs to sensory pathways in the spinal cord. Prog Brain Res, 67: 207-225.
- Zhuo M (2009). Plasticity of NMDA receptor NR2B subunit in memory and chronic pain. Mol Brain 2: 4.
- Wang GS, Hong C J, Yen TY, Huang HY, Ou Y, et al. (2004) Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron 42: 113-128.
- Ruscheweyh R, Goralczyk A, Wunderbaldinger G, Schober A, Sandkuhler J (2006) Possible sources and sites of action of the nitric oxide involved in synaptic plasticity at spinal lamina I projection neurons. Neuroscience 141: 977-988.
- Lin S Y, Wu K, Levine ES, Mount HT, Suen PC, et al. (1998) BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res 55: 20-27.
- Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, et al. (2004) APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem 279: 53379-53386.
- Bley KR, Hunter JC, Eglen RM, Smith JA (1998) The role of IP prostanoid receptors in inflammatory pain. Trends Pharmacol Sci 19: 141-147.
- Woolf C J, Wall PD (1986) Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. J Neurosci 6: 1433-1442.
- Michael G J, Averill S, Nitkunan A, Rattray M, Bennett DL, et al. (1997) Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci 17: 8476-8490.
- Fields RD, Eshete F, Stevens B, Itoh K (1997) Action potential-dependent regulation of gene expression: temporal specificity in ca2+, cAMP-responsive element binding proteins, and mitogen-activated protein kinase signaling. J Neurosci 17: 7252-7266.
- Ren K Ruda MA (1994) A comparative study of the calcium-binding proteins calbindin-D28K, calretinin, calmodulin and parvalbumin in the rat spinal cord. Brain Res Brain Res Rev 19: 163-179.
- Meng CF, Zhu XJ, Peng G, Dai DQ (2007) Re-expression of methylation-induced tumor suppressor gene silencing is associated with the state of histone modification in gastric cancer cell lines. World J Gastroenterol 13: 6166-6171.
- Sokolovz NE, Shiryaeva NV, Dyuzhikova NA, Savenko YN, Vaido AI. (2006). Effect of long-term mental and pain stress on the dynamics of H4 histone acetylation in hippocampal neurons of rats with different levels of nervous system excitability. Bull Exp Biol Med, 142(3), 341-343.
- Reiter RJ, Acuna-Castroviejo D, Tan DX, Burkhardt S (2001) Free radical-mediated molecular damage. Mechanisms for the protective actions of melatonin in the central nervous system. Ann N Y Acad Sci 939: 200-215.
- Acuna-Castroviejo D, Escames G, Reiter RJ (2006) Melatonin therapy in fibromyalgia. J Pineal Res 40: 98-99.
- Reiter RJ, Acuna-Castroviejo D, Tan DX (2007) Melatonin therapy in fibromyalgia. Curr Pain Headache Rep 11: 339-342.
- Rozen TD (2003). Melatonin as treatment for idiopathic stabbing headache. Neurology 61: 865-866.
- Song GH, Leng PH, Gwee KA, Moochhala SM, Ho KY (2005) Melatonin improves abdominal pain in irritable bowel syndrome patients who have sleep disturbances: a randomised, double blind, placebo controlled study. Gut 54: 1402-1407.
- Sutton MA, Wall NR, Aakalu GN, Schuman EM (2004) Regulation of dendritic protein synthesis by miniature synaptic events. Science, 304: 1979-1983.
- Carlson LE, Campbell TS, Garland SN, Grossman P (2007) Associations among salivary cortisol, melatonin, catecholamines, sleep quality and stress in women with breast cancer and healthy controls. J Behav Med 30: 45-58.
- Scheer F A, Zeitzer JM, Ayas NT, Brown R, Czeisler CA, et al. (2006) Reduced sleep efficiency in cervical spinal cord injury; association with abolished night time melatonin secretion. Spinal Cord 44: 78-81.
- Schmid DA, Wichniak A, Uhr M, Ising M, Brunner H, et al. (2006) Changes of sleep architecture, spectral composition of sleep EEG, the nocturnal secretion of cortisol, ACTH, GH, prolactin, melatonin, ghrelin, and leptin, and the DEX-CRH test in depressed patients during treatment with mirtazapine. Neuropsychopharmacology 31: 832-844.
- Chiechio S, Zammataro M, Morales, ME, Busceti CL, Drago F, et al. (2009) Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol 75: 1014-1020.
- Sharma R, Ottenhof T, Rzeczkowska PA, Niles LP (2008) Epigenetic targets for melatonin: induction of histone H3 hyperacetylation and gene expression in C17.2 neural stem cells. J Pineal Res 45: 277-284.
- Schule B, Albalwi M, Northrop E, Francis DI, Rowell M, et al. (2005) Molecular breakpoint cloning and gene expression studies of a novel translocation t(4;15)(q27;q11.2) associated with Prader-Willi syndrome. BMC Med Genet 6: 18.
- Sheedy F J, O'Neill LA (2008) Adding fuel to fire: microRNAs as a new class of mediators of inflammation. Ann Rheum Dis 67 Suppl 3: 50-55.
- Tili E, Michaille JJ, Costinean S, Croce CM (2008) MicroRNAs, the immune system and rheumatic disease. Nat Clin Pract Rheumatol 4: 534-541.
- Zhang J, Cheng H, Chen J, Yi F, Li W, et al. (2009) Involvement of activated astrocyte and microglia of locus coeruleus in cardiac pain processing after acute cardiac injury. Neurol Res 31: 432-438.
- Yang S N, Yu J, Mayr GW, Hofmann F, Larsson O, et al. (2001) Inositol hexakisphosphate increases L-type Ca2+ channel activity by stimulation of adenylyl cyclase. FASEB J 15: 1753-1763.
- Yamamoto M, Ueda R, Takahashi K, Saigo K, Uemura T (2006) Control of axonal sprouting and dendrite branching by the Nrg-Ank complex at the neuron-glia interface. Curr Biol 16: 1678-1683.
- Abramson SB (2008) Nitric oxide in inflammation and pain associated with osteoarthritis. Arthritis Res Ther 10: S2.
- Blanc V, Henderson JO, Newberry RD, Xie Y, Cho SJ, et al. (2007) Deletion of the AU-rich RNA binding protein Apobec-1 reduces intestinal tumor burden in Apc(min) mice. Cancer Res 67: 8565-8573.
- Bostrom K, Garcia Z, Poksay KS, Johnson DF, Lusis AJ, et al. (1990) Apolipoprotein B mRNA editing. Direct determination of the edited base and occurrence in non-apolipoprotein B-producing cell lines. J Biol Chem 265: 22446-22452.
- Bozsik A, Kokeny S, Olah E (2007) Molecular mechanisms for the antitumor activity of inositol hexakisphosphate (IP6). Cancer Genomics Proteomics 4: 43-51.
- Bottai D, Guzowski JF, Schwarz MK, Kang SH, Xiao B, et al. (2002) Synaptic activity-induced conversion of intronic to exonic sequence in Homer 1 immediate early gene expression. J Neurosci 22: 167-175.
- Bradbury EJ, King VR, Simmons LJ, Priestley JV, McMahon SB (1998) NT-3, but not BDNF, prevents atrophy and death of axotomized spinal cord projection neurons. Eur J Neurosci 10: 3058-3068.
- Bura SA, Nadal X, Ledent C, Maldonado R, Valverde O (2008) A 2A adenosine receptor regulates glia proliferation and pain after peripheral nerve injury. Pain 140: 95-103.
- Charbel Issa P, Lever IJ, Michael GJ, Bradbury EJ, Malcangio M (2001) Intrathecally delivered glial cell line-derived neurotrophic factor produces electrically evoked release of somatostatin in the dorsal horn of the spinal cord. J Neurochem 78: 221-229.
- Chamberlain SJ, Brannan CI (2001) The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics 73: 316-322.
- Didier-Bazes M, Chouaf L, Hardin H, Aguera M, Fevre-Montange M, et al. (1991) Developmental neuron-glia interaction: role of the serotonin innervation upon the onset of GABA uptake into the ependymocytes of the rat subcommissural organ. Brain Res Dev Brain Res 63: 135-139.
- Eyman M, Cefaliello C, Ferrara E, De Stefano R, Lavina ZS, et al. (2007) Local synthesis of axonal and presynaptic RNA in squid model systems. Eur J Neurosci 25: 341-350.
- Giuditta A, Chun JT, Eyman M, Cefaliello C, Bruno AP, et al. (2008) Local gene expression in axons and nerve endings: the glia-neuron unit. Physiol Rev 88: 515-555.
- Huang D, Yang CZ, Yao L, Wang Y, Liao YH, et al. (2008) Activation and overexpression of PARP-1 in circulating mononuclear cells promote TNF-alpha and IL-6 expression in patients with unstable angina. Arch Med Res 39: 775-784.
- Kang H, Schuman EM (1996) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273: 1402-1406.
- Jayakar SS, Dikshit M (2004) AMPA receptor regulation mechanisms: future target for safer neuroprotective drugs. Int J Neurosci 114: 695-734.
- LaRue RS, Jonsson SR, Silverstein KA, Lajoie M, Bertrand D, et al. (2008) The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol 9: 104.
- Larsson O, Barker CJ, Sjoholm A, Carlqvist H, Michell RH, et al. (1997) Inhibition of phosphatases and increased Ca2+ channel activity by inositol hexakisphosphate. Science 278: 471-474.
- Mac Ginnitie A J, Anant S, Davidson NO (1995) Mutagenesis of apobec-1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme, reveals distinct domains that mediate cytosine nucleoside deaminase, RNA binding, and RNA editing activity. J Biol Chem 270: 14768-14775.
- Mehler MF, Mattick JS (2007) Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev 87: 799-823.
- Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10: 23-36.
- Milligan ED, Sloane E, Watkins LR (2008) Glia in pathological pain: a role for fractalkine. J Neuroimmunol 198: 113-120.
- Navaratnam N, Sarwar R (2006). An overview of cytidine deaminases. Int J Hematol 83: 195-200.
- Okuse K (2007) Pain signalling pathways: from cytokines to ion channels. Int J Biochem Cell Biol 39: 490-496.
- Petit V, Guetard D, Renard M, Keriel A, Sitbon M, et al. (2009) Murine APOBEC1 is a powerful mutator of retroviral and cellular RNA in vitro and in vivo. J Mol Biol 385: 65-78.
- Polese D, De Serpis AA, Ambesi-Impiombato A, Muscettola G, De Bartolomeis A (2002) Homer 1a gene expression modulation by antipsychotic drugs: involvement of the glutamate metabotropic system and effects of D-cycloserine. Neuropsychopharmacology 27: 906-913.
- Roloff AM, Anderson GR, Martemyanov KA, Thayer S A (2010) Homer 1a gates the induction mechanism for endocannabinoid-mediated synaptic plasticity. J Neurosci 30: 3072-3081.
- Prochnow C, Bransteitter R, Chen XS (2009) APOBEC deaminases-mutases with defensive roles for immunity. Sci China C Life Sci 52: 893-902.
- Ren K, Dubner R (2008) Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anaesthesiol 21: 570-579.
- Salter MW (2005) Cellular signalling pathways of spinal pain neuroplasticity as targets for analgesic development. Curr Top Med Chem 5: 557-567.
- Sawyer SL, Emerman M, Malik HS (2004) Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol 2: 275.
- Ryan TJ, Emes RD, Grant SG, Komiyama NH (2008). Evolution of NMDA receptor cytoplasmic interaction domains: implications for organisation of synaptic signalling complexes. BMC Neurosci 9: 6.
- Tappe A, Klugmann M, Luo C, Hirlinger D, Agarwal N, et al. (2006) Synaptic scaffolding protein Homer1a protects against chronic inflammatory pain. Nat Med 12: 677-681.
- Van Den Broeke, EN, Van Rijn, CM, Biurrun Manresa, et al. (2010) Neurophysiological Correlates of Nociceptive Heterosynaptic Long-Term Potentiation in Humans. J Neurophysiol 103: 4
- Watrin F, Le Meur E, Roeckel N, Ripoche MA, Dandolo L, (2005). The Prader-Willi syndrome murine imprinting center is not involved in the spatio-temporal transcriptional regulation of the Necdin gene. BMC Genet 6: 1.
- Wang YJ, Wang X, Zhang H, Zhou L, Liu S, et al. (2009) Expression and regulation of antiviral protein APOBEC3G in human neuronal cells. J Neuroimmunol 206: 14-21.
- Wang Y, Bergmeier LA, Stebbings R, Seidl T, Whittall T, et al. (2009) Mucosal immunization in macaques upregulates the innate APOBEC 3G anti-viral factor in CD4(+) memory T cells. Vaccine 27: 870-881.
- Yu XM, Askalan R, Keil GJ, Salter MW (1997) NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275: 674-678.
- Zhang C, An J, Strickland DK, Yepes M (2009) The low-density lipoprotein receptor-related protein 1 mediates tissue-type plasminogen activator-induced microglial activation in the ischemic brain. Am J Pathol 174: 586-594.
- Wilson-Gerwing TD, Dmyterko MV, Zochodne DW, Johnston JM, & Verge VM (2005) Neurotrophin-3 suppresses thermal hyperalgesia associated with neuropathic pain and attenuates transient receptor potential vanilloid receptor-1 expression in adult sensory neurons. J Neurosci 25: 758-767.
- Taylor AM, Berchtold NC, Perreau VM, Tu CH, Li Jeon N, et al. (2009) Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci 29: 4697-4707.
- Sittl R, Boujong D, Griessinger N (1996) [Physio- and pathophysiology of acute pain with special consideration of pain prevention]. Anaesthesist 45 Suppl 3: 72-73.
- Mukhopadhyay D, Anant S, Lee RM, Kennedy S, Viskochil D, et al. (2002) C-->U editing of neurofibromatosis 1 mRNA occurs in tumors that express both the type II transcript and apobec-1, the catalytic subunit of the apolipoprotein B mRNA-editing enzyme. Am J Hum Genet 70: 38-50.
- Otoshi K, Kikuchi S, Konno S, Sekiguchi M (1970) The reactions of glial cells and endoneurial macrophages in the dorsal root ganglion and their contribution to pain-related behavior after application of nucleus pulposus onto the nerve root in rats. Spine 35: 10-17.
- Diagana TT, Thomas U, Prokopenko SN, Xiao B, Worley PF, et al. (2002) Mutation of Drosophila homer disrupts control of locomotor activity and behavioral plasticity. J Neurosci 22: 428-436.

