Keywords
Ethnic differences; Pain; Capsaicin; Naloxone; Endogenous opioids
Introduction
Ethnic differences in the prevalence, severity, and impact of a number of clinical pain conditions have been well documented [1,2]. Black pain patients generally display greater pain-related symptoms, suffering, pain interference, and disability relative to White patients [1,3]. Similarly, Black patients report higher levels of pain unpleasantness, greater emotional responses to pain, increased pain behaviors, and may not receive the same benefit from treatment as their White counterparts [1]. These differences are mirrored in the laboratory, with Black participants reporting an enhanced sensitivity to a variety of evoked pain testing methods when compared to White participants. Black participants report greater sensitivity (i.e., a lower pain threshold) to a variety of quantitative sensory testing (QST) protocols including thermal pain, cold pressor pain, ischemic pain and electrical stimulation, as well as greater temporal summation and reduced conditioned pain modulation (CPM) [2,4-11]. One mechanism theorized to contribute to these disparities is a differential functioning of the endogenous pain-regulatory systems between groups. For example, Black participants display reduced CPM in comparison to White participants [9,11,12]. CPM occurs when one painful stimulus inhibits the perception of a second simultaneous pain stimulus to a distant body site, and may be influenced by functioning of the endogenous opioid system [13,14].
Endogenous opioid analgesia derives in part from endogenous ligands of the mu opioid receptor (beta-endorphin), the same receptor activated by the most clinically-used opioid analgesic medications. Beta-endorphin, which is released in response to stressful and painful stimuli, is known to have analgesic effects, and can influence responses to both evoked laboratory pain stimuli and clinical pain [15-18]. One way to examine individual differences in functioning of the endogenous opioid system is to compare pain responses after placebo administration to pain responses after receiving naloxone, an opioid receptor antagonist which temporarily blocks the effects of endogenous opioids. Naloxone has been used in the clinical setting to block or reverse the effects of opioid agents. Naloxone, at doses of approximately 0.1 mg/kg, has also been used in conjunction with experimental-elicited pain stimuli as a means of further examining the function of endogenous opioids [19-21]. The current study administered a high dose of naloxone, in a placebo-controlled, double-blind, randomized fashion to evaluate group differences in endogenous opioid analgesic system function. As naloxone has been shown to increase laboratory pain responsiveness in those with effectively functioning endogenous opioid systems, we hypothesized that the NHW participants would display an increase in pain responsiveness following naloxone administration and that NHB participants’ pain would be unchanged by naloxone (i.e., NHB would show lower EO analgesia than NHW participants) [19,22].
Methods
The current secondary data analysis examined 39 healthy participants that were a part of a larger randomized, double blind, placebo-controlled crossover experimental study focused on distraction analgesia.
Participants were recruited via flyers posted around the community and by word-of-mouth. Participants completed one in-person screening and four experimental sessions, lasting up to two hours each, which involved the application of capsaicin under non-distraction conditions and distraction conditions (to be detailed elsewhere). In each of the four sessions, capsaicin was applied (as described below) and rated every five min. Similar to previous studies, naloxone (0.1 mg/kg) or placebo (saline) was administered after 25 min, when capsaicin pain is known to peak [19,21,22]. The current secondary analyses include the non-distraction conditions, with double-blind crossover administration of either naloxone or placebo in separate sessions (within subject) as described below.
Participants
Participants were asked to self-identify their race (Table 1). Only those participants who identified their ethnic/racial background as Black/African American (NHB) or NHW were included in the study. Participants were excluded if they reported serious medical conditions, chronic pain syndromes, current alcohol or drug abuse problems, were taking opioid analgesics for pain, or were unable to perceive or tolerate capsaicin procedures (described in the in-person screening). The study was conducted in accordance with the Helsinki declaration and the study protocol was approved by the Johns Hopkins Institutional Review Board. Written informed consent was obtained from all participants before the study.
| Variable (n) |
Non-Hispanic Black (19) |
Non-Hispanic White (20) |
| Sex (%female) |
52.6% |
50.0% |
| Age (SD) |
26.4 (6.7) |
24.9 (3.4) |
| BMI (SD) |
25.2 (5.2) |
24.2 (3.0) |
Education*
High School/GED
> Some college |
21.1%
78.9% |
0%
100% |
| Average Pain Ratings-Saline (30-90 min) |
53.4 (25.7) |
38.3 (21.9) |
| Average Pain Ratings-Saline during peak effects (40-65 min) |
52.7 (25.1) |
38.58 (21.9) |
| Average Pain Ratings-Naloxone (30-90 min) |
42.4 (21.3) |
37.2 (25.3) |
| Average Pain Ratings-Naloxone during peak effects (40-65 min) |
41.9 (20.4) |
37.7 (26.3) |
Table 1: Demographic and session data by group. Post drug administration average pain ratings are presented (30-90 min) as well as peak drug effect ratings (40-65 min).
In-person screening
Following a telephone screening, eligible participants were scheduled for an in-person screening session. Upon arrival, verbal and written informed consent was obtained, and then a urine screen to confirm absence of recent opioid use (to avoid precipitating acute withdrawal upon naloxone administration) and pregnancy test were performed. Participants completed a health history questionnaire and several psychological questionnaires. Participants then underwent capsaicin procedures (described below) for 30 min to ensure eligibility. Participants who reported no pain or negligible levels of capsaicin induced pain (<20 NHB and 6 NHW]) or very high levels of capsaicin induced pain (e.g. >80 [9 NHB and 2 NHW]) on a 0-100 scale (0 being “no pain at all” and 100 being “the most intense pain imaginable”) were excluded from the study to avoid floor and ceiling effects in the primary distraction study. Participants were then scheduled to undergo one of four psychophysical testing sessions, in a randomized order, with each scheduled at least 1 week apart [14]. Only the two non-distraction conditions are described here.
Capsaicin procedure
Capsaicin procedures were conducted similar to previous reports and included application of a thick piece of non-porous dressing to the skin at one of two randomized dorsal nondominant hand locations [22-27]. This dressing was a template for cream application and included a 6.25 cm2 hole cut into the center of it (used to standardize the area of application). Approximately 0.35 g of 10% capsaicin cream was applied inside this hole and evenly spread on the skin. The area was then covered by TegadermTM transparent dressing (3M Health Care, St. Paul, MN, USA). Pain induced by topical capsaicin varies strongly as a function of skin temperature, thus a peltierdevice heating element (Medoc US, Minneapolis, MN, USA) was strapped onto the area [28]. This device was held at a constant temperature of 40°C during the session. Following completion of each session, the capsaicin cream was removed from the skin.
Capsaicin with drug sessions
Upon arrival, participants were seated comfortably and an intravenous line was established in the non-dominant arm. After a 30 min wait, capsaicin and heat were applied as described previously. At the 25 min mark in each session, the assigned study drug (either naloxone or saline [D5W], stored and dispensed by the Johns Hopkins Bayview Medical Center Pharmacy and administered by the Clinical Research Unit) was pushed over three to five min following a double-blind, randomized design (Figure 1). Naloxone’s measured serum half-life in humans is approximately 60 min, with peak drug activity occurring approximately 15 min after administration. After the study drug randomly assigned for that session was administered, participants rated their pain using the 0-100 pain intensity scale every 5 min for 60 min (90 min total for the capsaicin and drug procedures). In the second session, identical procedures were followed but with administration of the other study drug.
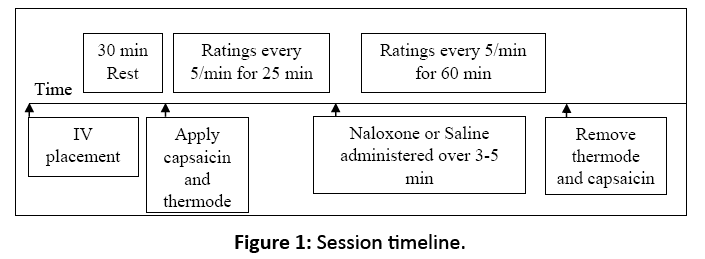
Figure 1: Session timeline.
Data analysis
All analyses were conducted using the SPSS statistical package version 23. Preliminary analyses included chisquared and t-tests to investigate ethnic group (NHB versus NHW participants) differences on various measures including demographic characteristics and pain. As an index of EO function, opioid blockade effects were derived for each participant by subtracting the mean post drug pain responses in the placebo condition from comparable mean ratings following naloxone administration (i.e., mean of ratings every 5 min from min 40-65; the window of maximal naloxone activity). Blockade effects were derived such that higher positive scores reflect greater EO analgesic function [15]. Larger negative scores indicate paradoxical analgesia with naloxone. Any demographic measure that differed between groups was included as a covariate in an analysis of covariance (ANCOVA) to evaluate ethnic differences in EO function (the primary analysis). Average pain during the saline session prior to drug administration was calculated and also used as a covariate in analyses.
Results
Demographic information for both groups is presented in Table 1. Of the demographic variables, only education differed between the two groups (p=0.047) and was thus included as a covariate in further analyses along with pre-drug pain ratings, which did not differ by group (p>0.05). Average pain ratings (30-90 min) in the placebo condition did not differ significantly (p=0.09) between groups when controlling for education, but were in the expected direction (NHB saline pain: x?=53.7 [25.8]; NHW saline pain: x?=38.3 [21.9]). In the naloxone condition, pain ratings also did not differ significantly (p=0.44) between NHB and NHW participants (NHB naloxone pain: x?=42.4 [21.3], NHW naloxone pain: x?=37.2 [25.3]).
Opioid blockade effects
A group difference in blockade effects emerged (F[1, 36]=5.4, p=0.038), with NHB participants displaying significantly lower blockade effects (mean=-10.8, SD=10.1) compared to NHW participants (mean=-0.89, SD=11.5) (Figure 2). This difference appears driven by NHB participants’ significant reduction in pain in the naloxone condition relative to placebo, elucidated by a one-sample t-test separately in each group (paradoxical naloxone analgesia; p=<0.001), an effect not significant in the NHW group (p=0.73). Thirty five percent of the NHW participants displayed the hypothesized positive blockade effects indicating EO analgesia (i.e., an increase in pain with naloxone), while only 10% of the NHB participants exhibited evidence of EO analgesia).
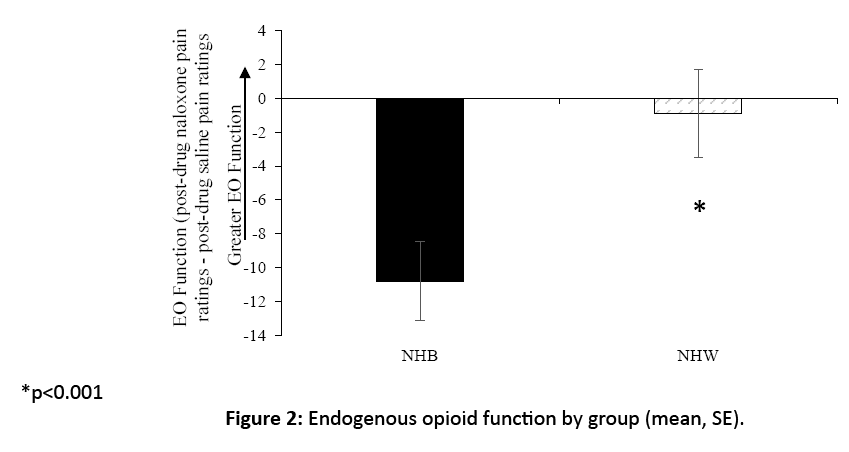
Figure 2: Endogenous opioid function by group (mean, SE).
Discussion
Ethnic disparities in clinical pain and laboratory-induced pain responses are well established [1,2,29,30]. Several factors such as biological, psychological, and sociocultural mechanisms contribute to differences in pain prevalence, perception, and reporting. Using multiple pain stimulus modalities in laboratory settings, NHB participants, when compared to NHW’s, report greater pain intensity and display lower pain tolerance to these stimuli. Furthermore, studies show that reduced tolerance of pain in NHB participants could be partially attributable to differential functioning in endogenous pain modulatory systems [1,9,11]. It is important to note that although there are robust clinical and laboratory differences in pain perception between NHW and NHB participants, the factors that lead to these differences have yet to be elucidated. There is more genetic variability within groups than between groups. Differences between ethnic groups may arise with different social or cultural experiences in early childhood and early experiences appear to have the ability to alter one’s biology. The hypothalamic-pituitary-adrenal (HPA) axis is altered with early stress exposure and a person’s central nervous system may adapt as a result [31-33].
The findings of the current study are consistent with their being differences in endogenous pain modulatory system function between NHB and NHW participants, although the nature of those differences was somewhat surprising. NHB participants reported a significant pain reduction during the naloxone condition (relative to placebo), an effect not observed in NHW participants, suggesting prominent naloxone-induced analgesia that was restricted to the NHB group. These findings are somewhat counter-intuitive to the typical effects of naloxone given at doses similar to the current study on both laboratory evoked pain and clinical pain [19,29,34]. Naloxone, by blocking endogenous opioid activity that contributes to endogenous pain modulation, increases laboratory pain in those with effectively functioning endogenous opioid systems. We hypothesized that elevated evoked pain responsiveness observed in prior work, if deriving in part from differences in endogenous opioid analgesia, would be reflected in NHB participants demonstrating impaired endogenous opioid functioning as compared to NHW participants. We thus expected that the NHW participant’s pain would increase with naloxone, while NHB participant’s pain would stay roughly unchanged. Although EO analgesia (positive blockade effects) tended to be more common in NHW than in NHB participants as expected, the ethnic differences observed in this study appeared driven primarily by the paradoxical analgesia observed in the NHB group.
While prior work in this area has calculated opioid blockade effects as the increase in pain that occurs following administration of naloxone or naltrexone, it is clear that a significant proportion of participants in these studies experience paradoxical naloxone analgesia or hypoalgesia: that is, a reduction in pain following administration of a competitive mu receptor antagonist. That opioid antagonists, even at high doses as in the current study, may in some cases have notable paradoxical analgesic effects have been reported previously [12]. Foar example, a substantial minority of patients with chronic low back pain showed paradoxical analgesia following IV naloxone administration, and an even larger subset of participants in a large study (n=151) of healthy young adults showed paradoxical hypoalgesic responses after oral administration of naltrexone [12,29]. While the mechanisms subserving individual differences in response to endogenous opioid blockade have not been definitively identified, it has been hypothesized that hypoalgesic effects of competitive opioid antagonists could be due to the inhibitory effects on presynaptic autoreceptors which are responsible for down-regulation of opioid release [12,35]. That is, if individuals have tonic hyper-activation of autoreceptors, resulting in reduced opioid release into the synaptic cleft and consequent hypersensitivity to noxious stimuli, then antagonism of autoreceptor activation may “unmask” a previously-inhibited endogenous opioid analgesia. While this hypothesis is speculative at present, a greater preponderance of autoreceptor activation in NHB participants could help explain the present pattern of findings. It should also be noted that our finding of paradoxical naloxone analgesia is consistent with clinical studies utilizing an oral opioid antagonist, naltrexone, although these studies were not conducted in NHB or predominantly Black patient groups [36,37]. Younger and colleagues documented that fibromyalgia patients receiving low-dose naltrexone treatment experienced a larger reduction in clinical pain ratings compared to placebo. A key methodological distinction, however, between our work and the clinical studies is the low dose in the latter studies and the administration method for the opioid antagonist (3.0-4.5 mg of oral naltrexone compared to the typical clinical dose of 50mg oral, vs. our use of naloxone) [36,37].
Potential mechanisms for the paradoxical analgesic effects of naloxone observed in NHB participants in the current study warrant further investigation, as they might contribute to racial differences in pain responsiveness. One possibility is that functional genetic mutations in the mu opioid receptor might alter naloxone responses (Rs. 563649) and be associated with a more pain sensitive phenotype [38]. To the extent that such mutations may differ in prevalence by race, they might contribute to the observed pattern of results. It is also conceivable that altered responsiveness to naloxone in NHB participants might be related to group differences in neuroinflammatory mechanisms that help contribute to a more pain sensitive phenotype. Limited data suggests that the opioid antagonist naloxone also acts on glial cells via regulation of the neuroimmune pathway [36,37]. In both clinical and experimental settings, inflammation has been shown to play a significant role in chronic pain.
There have been few studies conducted to investigate the effects of high-dose naloxone (0.1 mg/kg) on capsaicin-elicited pain perception. Within the current study, there was both a lack of expected significant positive blockade effects in the NHW and NHB groups and significant pain reductions during the naloxone session in NHB participants. A limitation of the study is that it did not use any manipulations (e.g. CPM) designed to trigger endogenous analgesic mechanisms which may have elucidated these unexpected results. An additional limitation of the study is the small sample size of NHB and NHW participants, which hinders generalization of the hypothesis of ethnic disparities in endogenous pain regulatory function. In future studies, these preliminary data will be strengthened by acquiring larger groups of both NHB and NHW participants. Furthermore, a follow-up study will allow for in-depth exploration of other individual characteristics beyond ethnicity, including genetic variation(s) of the particular signaling pathway, which may affect the function of endogenous neuromodulators in response to naloxone.
There is the possibility that the paradoxical effect we noted might be viewed not as reflecting differences in endogenous pain modulation, but rather as reflecting a previously undescribed racial difference in pharmacological responses to naloxone. If true, this would have methodological implications for interpreting studies (particularly those with large NHB participant samples) using opioid blockade to probe endogenous opioid systems.
Moving forward, continuing to explore the effects of EO function using naloxone/placebo challenges in order to understand the mechanism(s) involved in endogenous pain regulatory pathways among ethnic groups is an important undertaking. Utilization of PET imaging will presumably allow researchers to identify specific areas of the brain involved in the complex interplay of hyperalgesia and pain modulation between the CNS and PNS. Upon employing PET imaging, we speculate that the current results might suggest that a decrease in pain after administering naloxone in NHB might relate to an increase in the algesic threshold through the inhibition of microglial cell activation and its subsequent secretion of proinflammatory cytokines. The connection between these two quintessential factors will shed light on particular neural pathways on which naloxone acts.
There are additional limitations that should be considered when evaluating the results of the current study. By only including NHB and NHW participants, we are limited in understanding other racial/ethnic groups (e.g. Asian populations); additionally, no within group heterogeneity was explored, which may be larger than the group differences evaluated here. Future studies should offer more options for self-identification, so as to capture additional ethnic identifications. It also should be noted that by using the racial/ethnic category “Black or African American”, individuals who identify as Black but not African American may have been artificially grouped together. While the current methods were modeled after a previous study, we did not have participants engage in an activity during capsaicin testing [19]. This, along with any unknown minor methodological differences could have influenced results and conceivably contributed to the difference in our findings. Additionally, by eliminating “low capsaicin pain” subjects a priori, this may have reduced the observed EO effect, potentially reducing our ability to properly evaluate the full spectrum of endogenous opioid analgesia. Finally, future work should extend these analyses from healthy participants, whose generalizability is limited, into chronic pain populations. Further research should also continue to evaluate possible sources of variability in pain perception and report, with the objective of eliminating health disparities.
Conclusion
The findings of the current study suggest differences in endogenous pain modulatory system function between NHB and NHW participants, although the nature of those differences was somewhat surprising. NHB participants reported a significant pain reduction during the naloxone condition as compared to NHW participants, suggesting paradoxical naloxone-induced analgesia selective to the NHB group.
Acknowledgement
The authors would like to thank the Blaustein Pain Foundation, the JHBMC Clinical Research Unit and acknowledge additional members of our team who primarily conducted data collection, including Mpepera Simango, Kasey Bond, Lea McCauley, Sara Bounds and Sasha Beselman for helping with the administration, data collection, data entry and pharmacy requirements of the study. Finally, the subjects participating should be acknowledged, without their participation this study would not have been possible.
Funding Sources
This research was supported by grants K23-NS070933 (CMC), R01-MD009063 (CMC), R01-DA037891 (SB), K23- DA029609 (DAT), The Blaustein Pain Foundation, & The JHBMC Clinical Research Unit.
References
- Campbell CM, Edwards RR (2012) Ethnic differences in pain and pain management. Pain Manag. 2:219-230.
- Campbell CM, Edwards RR, Fillingim RB (2005) Ethnic differences in responses to multiple experimental pain stimuli. Pain. 113:20-26.
- Edwards RR, Moric M, Husfeldt B, Buvanendran A, Ivankovich O, et al. (2005) Ethnic similarities and differences in the chronic pain experience: A comparison of African American, Hispanic and white patients. Pain Med. 6:88-98.
- Riley JL, Wade JB, Myers CD, Sheffield D, Papas RK, et al. (2002) Racial/ethnic differences in the experience of chronic pain. Pain. 100:291-298.
- Chapman WP, Jones CM (1944) Variations in cutaneous and visceral pain sensitivity in normal subjects. J Clin Invest. 23:81-91.
- Edwards RR, Doleys DM, Fillingim RB, Lowery D (2001) Ethnic differences in pain tolerance: Clinical implications in a chronic pain population. Psychosom Med. 63:316-323.
- Sheffield D, Biles PL, Orom H, Maixner W, Sheps DS (2000) Race and sex differences in cutaneous pain perception. Psychosom Med. 62:517-523.
- Walsh NE, Schoenfeld L, Ramamurthy S, Hoffman J (1989) Normative model for cold pressor test. Am J Phys Med Rehabil. 68:6-11.
- Campbell CM, France CR, Robinson ME, Logan HL, Geffken GR, et al. (2008b) Ethnic differences in the nociceptive flexion reflex (NFR). Pain. 134:91-96.
- Mechlin B, Heymen S, Edwards CL, Girdler SS (2011) Ethnic differences in cardiovascular-somatosensory interactions and in the central processing of noxious stimuli. Psychophysiology. 48:762-773.
- Campbell CM, France CR, Robinson ME, Logan HL, Geffken GR, et al. (2008a) Ethnic Differences in Diffuse Noxious Inhibitory Controls (DNIC). J Pain. 9:759-766.
- France CR, al'Absi M, Ring C, France JL, Harju A, et al. (2007) Nociceptive flexion reflex and pain rating responses during endogenous opiate blockade with naltrexone in healthy young adults. Biol.Psychol. 75:95-100.
- Le Bars D, Chitour D, Kraus E, Dickenson AH, Besson JM, et al. (1980) Effect of naloxone upon diffuse noxious inhibitory controls (DNIC) in the rat. Brain Res. 204:387-402.
- Willer JC, Le Bars D, De Broucker T (1990) Diffuse noxious inhibitory controls in man: Involvement of an opioidergic link. Eur.J Pharmacol. 182:347-355.
- Bruehl S, Burns JW, Gupta R, Buvanendran A, Chont M, et al. (2013) Endogenous opioid function mediates the association between laboratory-evoked pain sensitivity and morphine analgesic responses. Pain. 154:1856-1864.
- Dalayeun JF, Nores JM, Bergal S (1993) Physiology of beta-endorphins. A close-up view and a review of the literature. Biomed Pharmacother. 47:311-320.
- Spetea M (2013) Opioid receptors and their ligands in the musculoskeletal system and relevance for pain control. Curr Pharm Des. 19:7382-7390.
- Sprouse-Blum AS, Smith G, Sugai D, Parsa FD (2010) Understanding endorphins and their importance in pain management. Hawaii Med J. 69:70-71.
- Anderson WS, Sheth RN, Bencherif B, Frost JJ, Campbell JN, et al. (2002) Naloxone increases pain induced by topical capsaicin in healthy human volunteers. Pain. 99:207-216.
- Bruehl S, Chung OY, Ward P, Johnson B (2004) Endogenous opioids and chronic pain intensity: interactions with level of disability. Clin J Pain. 20:283-292.
- Bruehl S, Chung OY (2006) Parental history of chronic pain may be associated with impairments in endogenous opioid analgesic systems. Pain. 124:287-294.
- Bencherif B, Fuchs PN, Sheth R, Dannals RF, Campbell JN, et al. (2002) Pain activation of human supraspinal opioid pathways as demonstrated by [11C]-carfentanil and positron emission tomography (PET). Pain. 99:589-598.
- Campbell CM, Edwards RR, Carmona C, Uhart M, Wand G, et al. (2009) Polymorphisms in the GTP cyclohydrolase gene (GCH1) are associated with ratings of capsaicin pain. Pain. 141:114-118.
- Campbell CM, Bounds SC, Simango MB, Witmer KR, Campbell JN, Edwards, et al. (2010a) Self-reported sleep duration associated with distraction analgesia, hyperemia and secondary hyperalgesia in the heat-capsaicin nociceptive model. Eur J Pain. 15:561-567.
- Campbell CM, Quartana PJ, Buenaver LF, Haythornthwaite JA, Edwards RR (2010b) Changes in situation-specific pain catastrophizing precedes changes in pain report during capsaicin pain: A cross-lagged panel analysis among healthy, pain-free participants. J Pain. 11:876-884.
- Campbell CM, Witmer K, Simango M, Carteret A, Loggia ML, et al. (2010c) Catastrophizing delays the analgesic effect of distraction. Pain. 149:202-207.
- Campbell CM, Bounds SC, Simango MB, Witmer KR, Campbell JN, et al. (2011) Self-reported sleep duration associated with distraction analgesia, hyperemia and secondary hyperalgesia in the heat-capsaicin nociceptive model. Eur J Pain. 15:561-567.
- Dirks J, Petersen KL, Dahl JB (2003) The heat/capsaicin sensitization model: A methodologic study. J Pain. 4:122-128.
- Bruehl S, Burns JW, Gupta R, Buvanendran A, Chont M, et al. (2014) Endogenous opioid inhibition of chronic low-back pain influences degree of back pain relief after morphine administration. Reg Anesth Pain Med. 39:120-125.
- Rahim-Williams B, Riley JL III, Williams AK, Fillingim RB (2012) A quantitative review of ethnic group differences in experimental pain response: do biology, psychology and culture matter? Pain Med. 13:522-540.
- Burke NN, Finn DP, McGuire BE, Roche M (2017) Psychological stress in early life as a predisposing factor for the development of chronic pain: Clinical and preclinical evidence and neurobiological mechanisms. J Neurosci Res. 95:1257-1270.
- LaPrairie JL, Murphy AZ (2009) Neonatal injury alters adult pain sensitivity by increasing opioid tone in the periaqueductal gray. Front Behav Neurosci. 3:31.
- LaPrairie JL, Murphy AZ (2010) Long-term impact of neonatal injury in male and female rats: Sex differences, mechanisms and clinical implications. Front Neuroendocrinol. 31:193-202.
- Levine JD, Gordon NC, Fields HL (1979) Naloxone dose dependently produces analgesia and hyperalgesia in postoperative pain. Nature. 278:740-774.
- Kolaric S, Makulska-Nowak HE, Gumulka SW, Mizerska K (1999) Paradoxical effects of intracerebroventricular low-dose opioid antagonists in SHR with chronic pain. Life Sci. 65:395-402.
- Younger J, Noor N, McCue R, Mackey S (2013) Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, cross-over trial assessing daily pain levels. Arthritis Rheum. 65: 529-538.
- Younger J, Parkitny L, McLain D (2014) The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clin Rheumatol. 33:451-459.
- Shabalina SA, Zaykin DV, Gris P, Ogurtsov AY, Gauthier J, et al. (2009) Expansion of the human mu-opioid receptor gene architecture: novel functional variants. Hum Mol Genet. 18:1037-1051.