S. L. Balakrishna1, Bharti Gupta2 and Parikipandla Sridevi3*
1Model Rural Health Research Unit, ICMR, Badoni, Datia, MP, India
2Department of Biotechnology, Indira Gandhi National Tribal University, India
3Department of Biotechnology, Faculty of Science, Indira Gandhi National Tribal University, India
*Corresponding Author:
Dr. Parikipandla Sridevi
Assistant Professor, Department of Biotechnology, Faculty of Science
Indira Gandhi National Tribal University, Amarkantak
484887, Madhya Pradesh, India
Tel: +91-9630036673
E-mail: psridevi@igntu.ac.in
Received Date: May 22, 2018 Accepted Date: July 24, 2018 Published Date: July 31, 2018
Citation: Balakrishna SL, Gupta B, Sridevi P (2018) Expression Analysis of Cell Surface Markers: Cluster of Differentiation 4, Chemokine Receptor 5 and C-X-C Chemokine Receptor Type 4 in Different Cell Lines after Infection with HIV. J HIV Retrovirus Vol. 4 No.2:14 doi: 10.21767/2471-9676.100046
Keywords
HIV; CD4+ T cells; Cell surface markers; gp120; CD4; CCR5; CXCR4
Introduction
Human Immunodeficiency Virus (HIV) is a retrovirus that causes Acquired Immune deficiency Syndrome (AIDS). One of the principal cellular targets of HIV infection is the cluster differentiation 4 positive (CD4+) T lymphocytes [1]. HIV leads to the loss of T lymphocytes are a central factor in the progression of the disease, due to their central role in controlling immune responses. HIV infects T cells via interaction between glycoprotein gp120 and the CD4 molecule [2]. Infection of T cells is assisted by the interaction of T cells with C-X-C Chemokine receptor type 4 (CXCR4) and C-C Chemokine receptor type 5 (CCR5) co-receptors. The envelope of HIV-1 mediates virus entry into cells, which comprises surface gp120 glycoproteins non-covalently linked to transmembrane gp41 glycoprotein that/which embed the complex into the viral membrane. HIV-1 entry is initiated by gp120 binding to cellular CD4 [3], which is a high-affinity interaction that facilitates the initial attachment of virus to the target cell. The binding of gp120 to receptor CD4 results in a conformational change in gp120 which exposes the binding site for chemokine receptors CCR5 or CXCR4 [4,5].
Another study on CEM (lymphoblastic cells) and C8166 CD4+ T cells in vitro infection by HIV-1 virus effects on cellular gene expression on these different two cell lines reported in which the expression of 1506 cDNAs during HIV- 1 infection using cDNA microarrays and identified 20 differentially expressed genes that function in a variety of cellular pathways [6]. Out of the 20 genes 140 (RIP14000, T cell receptor α (TCRα) and Translocase of the Outer Membrane 34 (TOM 34) genes were found to be upregulated and prothymosin α and type IV protein tyrosine phospatase (type IV PTP) were downregulated [7].
Their study was the first quantitative large-scale analysis of host cell gene expression in HIV-1-infected cells and results supported the hypothesis that HIV-1 infection alters cellular gene expression at a global level which has not been recognized previously [8]. According to another cDNA analysis study human T cells upon HIV-1 infection showed that 15% of 2,000 cDNA were differentially expressed after infection, in which 27 markers were found to be significantly down-regulated and were up-regulated at peak HIV infection [9]. An in vitro study showed the level of mRNA in (Peripheral Blood Mononuclear Cells) PBMCs does not indicate the effects of HIV infection on the different cell populations that are present in the peripheral blood. PBMCs include monocytes, lymphocytes and circulating dendritic cells. The kinetics of virus replication depends on the stage of cellular differentiation at the time of virus infection [10]. CD4 (T helper) cells, the main target for HIVs, expression of the CD4 receptors and their destruction is associated with the deterioration of the immune system. T-lymphocytes do not support a productive infection with HIVs in vitro, unless they are first stimulated with agents such as mitogens or antigens. Activated T lymphocytes are easier to infect than non-activated ones [11].
In this study, we investigated the expression profile of cell surface markers in three types of cells infected with HIV-1 virus using quantitative RT-PCR analysis. Our study is an effort to fill the gap in knowledge of very early changes in host proteins after HIV infection at transcriptional level.
Materials & Methods
Cell and virus culture
SupT1 cells, Jurkat cells and PBMCs were propagated in RPMI (Gibco-BRL, Invitrogen, CA, USA) with 10% FBS (Gibco-BRL) at standard conditions of 5% CO2 and 37oC. HIV-1 93IN101 (subtype C) was cultured by infecting the SupT1 cells with cryopreserved viral stock in the presence of 8 μg/mL Polybrene (Sigma, St. Louis, MO, USA) overnight. The unbound virus was removed by washing the cells with 0.1M Phosphate Buffered Saline (PBS) three times and the cells were seeded back to complete medium. After 4 days of infection, the shedding virus was quantified in the culture supernatant with p24 ELISA kit (ABL Inc, Kensington, MD, USA), filtered using 0.45μ membrane filter (Pall Corporation, USA) and used for infection experiments. Infection was given at 20 ng (100 MOI) viruses for every 2x106 cells in the presence of 8 μg/mL Polybrene in serum free media.
HIV-1 p24 antigen assay
Cells were infected with HIV-1 for 2 h in the serum free media.
The unbound virus was washed with 0.1M PBS, three times and the cells were seeded back in the complete medium. After 4 days of infection, the shedding virus was quantified in the free culture supernatants with p24 ELISA kit following manufacturer’s instructions.
PBMCs isolation and culture
PBMCs were obtained from healthy volunteers as per Institutional Ethics Committee approval. PBMCs from healthy individuals were isolated and cultured from the blood drawn into heparin-coated tubes (Vacutainer; Becton Dickinson) as per standard protocol, with slight modifications. PBMCs were isolated by gradient centrifugation using Ficoll-Hypaque (Amersham Pharmacia). RPMI 1640 medium was used to re-suspend PBMCs and were cultured with PHA/IL-2 (2.5 μg/mL and 50 units/mL IL-2, respectively), supplemented with antibiotics penicillin (100 units/ mL) and streptomycin (100 μg/mL) along with Polybrene 2 μg/ mL. After 3 days of culturing, antibiotic was removed 24 h prior to harvest of cells and used for viral infections.
Viral infection
In all protein expression studies, cells were infected with live and heat inactivated virus separately. For each time point 5 x 106 cells were infected with HIV-1 and collected at 0, 1, 2, 4, 6 and 8 h of post infection.
Total RNA isolation and qRT-PCR
Total RNA from the cells was isolated using Trizol reagent (Sigma). The concentration of total RNA was assessed using NanoDrop (ND-1000, NanoDrop technologies, USA) spectrophotometer and the quality was checked by formaldehyde agarose gel electrophoresis First strand cDNA synthesis was carried out using superscript III (Invitrogen, USA) following manufacturer’s protocol. The primer sets used for qRT-PCR were designed using Primer Express software (Applied Biosystems, USA) such that at least one primer in each set flanked the intron-exon boundary to prevent amplification from genomic DNA (Table 1). All target genes analyzed and the endogenous control used in the present study (Gapdh). All qRT-PCRs were carried out in triplicates using power SYBR Green PCR master mix (Applied Biosystems) in an ABI-7500 fast real time PCR machine (Applied Biosystems) at an initial hold of 95oC (10 min) followed by a 2-step PCR reaction of 95oC (15 s) and 60oC (1 min) for 40 cycles according to the manufacturer’s protocol. Dissociation or melting curve analysis was performed for each gene to check for single amplification. During PCR, fluorescence accumulation resulting from DNA amplification was recorded using the ABI 7500 sequence detection system (SDS) software. Cycle threshold (Ct) values were obtained from the exponential phase of PCR amplification by SDS software. Gapdh was used as an endogenous control as it did not show any significant change in expression after infection. The target gene expression was normalized against gapdh expression taking o hour after infection as reference as per 2-ΔΔCT
| Name of Gene |
Amplicon |
Forward primer |
Reverse primer |
| GP120 |
204 bp |
5'- CAA TGT ATG CCC CTC CCA TCA GTG GAC -3' |
5'- CCT TGG TGG GTG CTA CTC CTA ATG GTT CA -3' |
| CD4 |
191 bp |
787 CTC CCG CTC CAC CTC ACC CTG 807 |
5'- CTC AGC ATC AGC TTA GGG GAG GTG G -3' |
| CCR5 |
183 bp |
5'- GAA GGT CTT CAT TAC ACC TGC AGC TCT C -3' |
5'- CCT GTG CCT CTT CTT CTC ATT TCG ACA C -3' |
| CXCR4 |
183bp Ref No seq: NM_003467 |
336 5'- CTG TCA GTG GCC GAC CTC CTC TTT GTC -3' 355 |
538 5'- GCT TCC TTG GCC TCT GAC TGT TGG TGG -3' 519 |
Table 1: The primer sequences used for qRT-PCR.
Statistical analysis
Data were presented as mean ± SEM (standard error of mean).
Number of samples/repeats for each experiment/assay is indicated in the legends to figures. All statistical analyses were done using the SPSS 10.0 software package (SPSS Inc., Chicago, IL, USA). Differences between groups were compared using Anova (single factor) test and for each test, P value of <0.05 (*) were considered significant.
Results
Primer sequences use for amplification of different genes is gives in Table 1. Primer specificity for each primer pair was confirmed by dissociation curve analysis given in Figure 1 and amplification plots of each gene given in Figure 2.
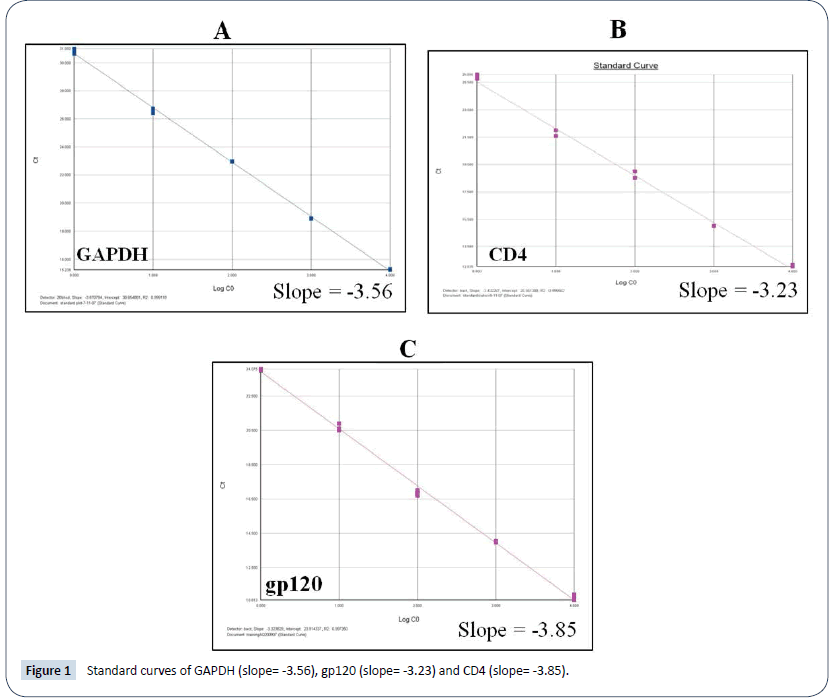
Figure 1: Standard curves of GAPDH (slope= -3.56), gp120 (slope= -3.23) and CD4 (slope= -3.85).
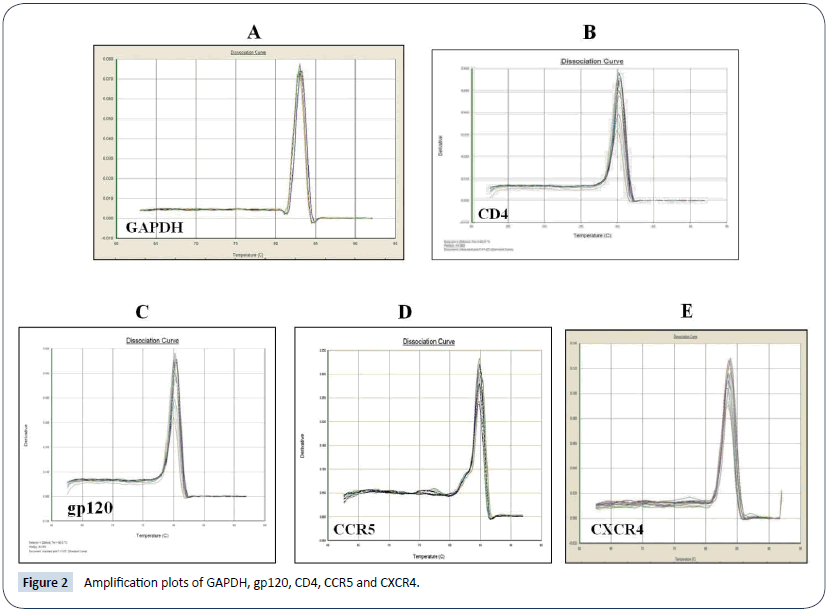
Figure 2: Amplification plots of GAPDH, gp120, CD4, CCR5 and CXCR4.
Expression of cells surface markers gp120, CD4, CCR5 and CXCR4 by qRT-PCR in infected cells at different points after HIV-1 infection in Jurkat cells
Expression analysis of different cell surface markers at different time points after HIV infection in Jurkat cells (Figure 3). Gp120 up-regulated at 1 h after HIV infection and showed a steady decrease from 2 h to 4 h. Both CD4 and CCR5 up regulated by 2 h of post infection in cell lines and decreased at later time points. CXCR4 showed a significant up regulation by 1 h in Jurkat cells after infection.
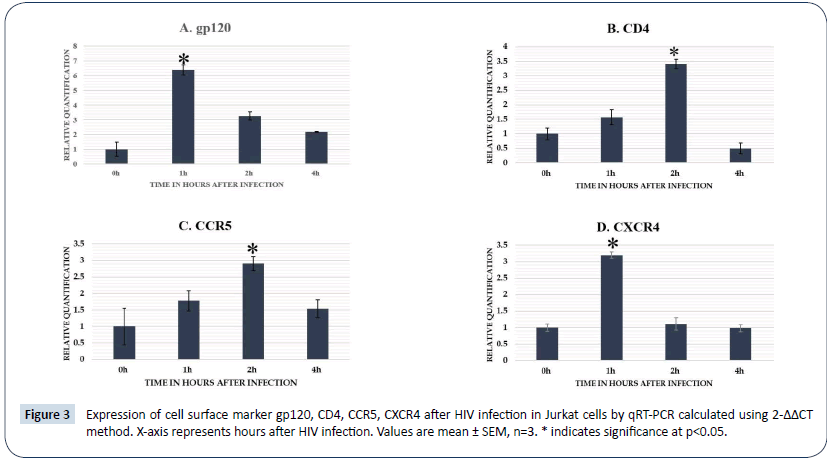
Figure 3: Expression of cell surface marker gp120, CD4, CCR5, CXCR4 after HIV infection in Jurkat cells by qRT-PCR calculated using 2-ΔΔCT
Expression of cells surface markers after HIV-1 infection in SupT1 cells
Expression analysis of different cell surface markers at different time points after HIV infection in SupT1 cells showed gp120 upregulated at 1 h after HIV infection and showed a steady decrease from 2 h to 4 h (Figure 4). Both CD4 and CCR5 up regulated by 2 h of post infection in the cell lines and decreased at later time points and CXCR4 showed a significant up regulation at 2 h post infection in SUP T1 cells.
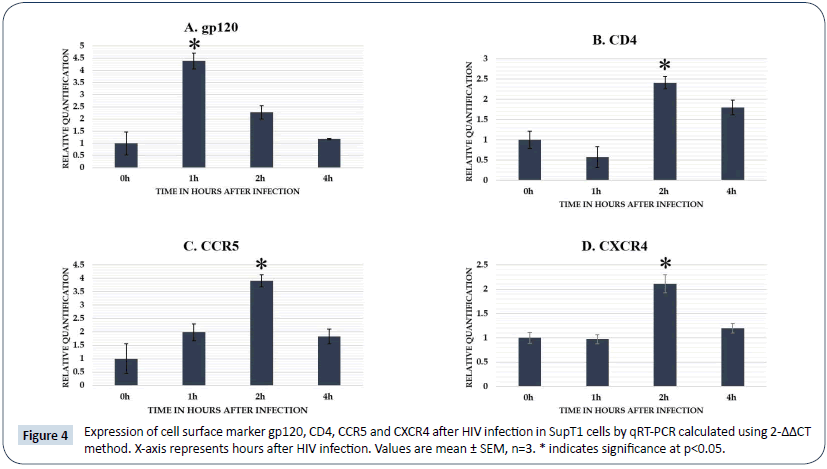
Figure 4: Expression of cell surface marker gp120, CD4, CCR5 and CXCR4 after HIV infection in SupT1 cells by qRT-PCR calculated using 2-ΔΔCT
Expression of cells surface markers after HIV-1 infection in PBMCs
Expression analysis of different cell surface markers at different time points after HIV infection in PBMCs (Figure 5). Gp120 and CD4 showed up-regulation at 1 h and 4 h post infection respectively in PBMCs but CCR5 and CXCR4 did not show significant difference in the expression at different time points after HIV infection.
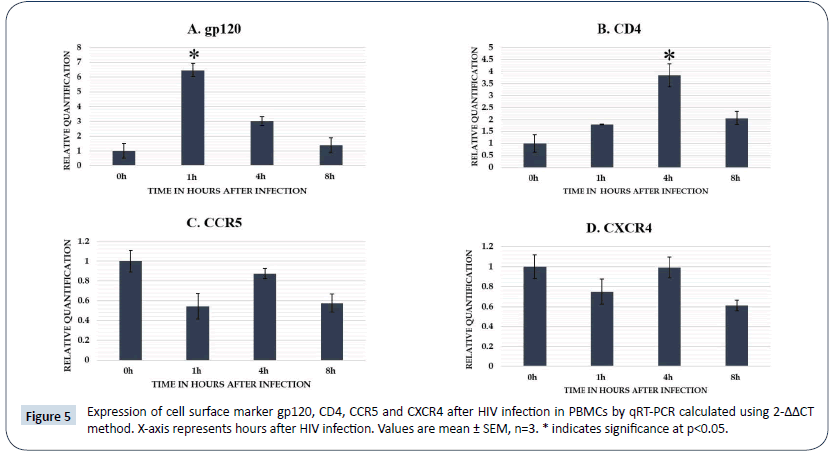
Figure 5: Expression of cell surface marker gp120, CD4, CCR5 and CXCR4 after HIV infection in PBMCs by qRT-PCR calculated using 2-ΔΔCT
Discussion
CCR5 and CXCR4 receptor interaction with the HIV-1 surface envelope glycoprotein gp120 triggers molecular events that eventually result in HIV infection. It has been shown that gp120 can induce rapid internalization of these receptors [12,13]. We studied this expression by using three different CD4+ T cell lines (Jurkat, SupT1 and PBMCs) and infected the cells with HIV-1 in vitro. Jurkat, SupT1 and PBMCs cells upon HIV-1 infection shows an up-regulation in gp120 and other surface receptor expression after 1 hpi, with a steady decrease in expression after 2 and 4 hpi in Jurkat and SupT1 cells. At early infection, the available transcripts of gp120 are from virions infected the host cell because the viral gene transcription occurs after 12 hpi only. There are several studies stating that, to prevent super infection and to escape from immune recognition the cell surface receptors of the host cell are generally degraded [14-16]. Hence, in this study we have used gp120 marker as an infection marker. Results show that viral infection occurred in our study till at 1 hpi. Later further infection of infected cells (super infection) may be restricted as well as the existing viral genomic RNA was reverse transcribed into cDNA or dsDNA. As a result of this, at later hpi the gp120 marker levels gradually decreased. The expression pattern of host cell surface markers correlates with earlier report that gp120 down-regulates the expression of both the surface receptors and CD4 in T Cell Depleted Marrow (TCDM) post infection of HIV, but our reports differ with the existing reports temporally i.e. instead of getting effects at 6-12 h after treatment, in our study we found these effects as early as 2 hpi. As reported previously [17] at 24 h infection the expression of CD4 remains low. Studies suggest that gp120-induced down-modulation of the CXCR4 and CD4 receptors may involve different mechanisms but that CXCR4 down-modulation induced by gp120 requires CD4 [17]. Our data supports this finding as in our study the CXCR4 and CCR5 receptors were observed to be up-regulated and this up-regulation dependent on the CD4 receptor. In PBMCs, we observed a similar up-regulation of gp120 and CD4 expression with no significant change in the CCR5 and CXCR4 expression. These studies provide new information about interaction of HIV- 1 envelope protein gp120 to the CD4 receptor and co-receptors CCR5 and CXCR4.
CD4, CCR5 and CXCR4 are cellular receptors for viruses are often down-regulated [18] from the plasma membrane following productive infection, making infected cells refractory to super infection by other viruses that use the same receptor for entry [19,20]. The decrease in surface expression may be caused in part by the formation of a complex between the viral receptor binding protein and cellular receptors in intracellular compartments [21]. Several mechanisms have been proposed to account for the down-regulation of CD4 following primate lentivirus infection [22]. Internalization of CD4 can occur upon binding of HIV-1 envelope glycoproteins [23]. In our study down-regulation of CD4 observed in SupT1cells. Followed by an up-regulation after 2 hpi and decreased at later time points.
Down-regulation of CD4 may also be mediated by the HIV-1 Nef and Vpu accessory proteins [24]. In CD4+ T cells there is an upregulation of CXCR4 expression after 1 hpi in Jurkat cells, CXCR4 up-regulation also observed in SupT1 cells after 2 h of HIV-1 post infection. Up-regulation of the CXCR4 co-receptor during productive infection by CD4-dependent HIV-1 strains was not observed in previous studies [25], thus, supporting our data.
CXCR4 is also down-regulated in cells infected with CD4- independent X4 HIV-1 isolate m7NDK [25]. HIV-1 cytopathic effects are responsible for the decrease in surface CXCR4 in RH9 and Jurkat cultures, CXCR4. CXCR4 is uniformly present on the cells in the RH9 and Jurkat cultures [26]. It is also possible that differences in the ability to down-regulate CXCR4 are cell specific. However, in current studies we observed that HIV-1 induces CXCR4 up-regulation in Jurkat and SupT1 cells lines 1 h and 2 h of post infection respectively we also observed a slight downregulation of CXCR4 in PBMCs after 1 hpi.
Alteration in CXCR4 [27] expression after infection by HIV-1 could result from sequestration of CXCR4 intracellular or from the direct effects of other HIV-1 proteins on the synthesis of CXCR4 or its transport to the cell surface. Several studies have shown that HIV-1 gp120 can displace chemokines from their receptors [28]. Interactions between gp120, CD4, and CXCR4 have also been well established [29]. Reduced levels of CXCR4 mRNA transcripts were observed in cells infected with CD4-independent HIV-1 isolate [25]. Furthermore, the modulation of CCR5 expression by the HIV viruses is at the level of transcription [30]. Further experiments will be needed to determine the mechanisms of down-modulation of surface CXCR4 by HIV-1.
In Indian patients elevated levels of CCR5 expressing target cells have reported, which may be due to abnormal immune activation from environmental sources. Increased expression of CCR5 on uninfected T cells may make them more susceptible to infection by CCR5-utilizing HIV-1 isolates, favoring selection and evolution of CCR5-utilizing isolates [31]. Our finding supports this study as we found an up-regulation of CCR5 receptor in both Jurkat and SupT1 cell lines after 1 and 2 hpi. Through this finding we can also propose that down-regulation of CCR5 after viral infection can prevent super infection and premature apoptosis of infected cells. It is therefore important to analyze CCR5 expression on different lymphocyte sub-populations.
Our results demonstrated that the release of the viral accessory proteins Vif, Vpr, and Nef which were enclosed with in the virion particle immediately after fusion can show regulatory effects in the host cell. The comparison of the viral infection in the form of gp120 transcripts showed nearly 2-fold decreased infection in Sup-T1 cells than others at 1 hpi. Hence, we can assume less availability of accessory proteins. Both Jurkat and Sup-T1 cells after infection showed increased transcripts of CD4 and CCR5 at 2 hpi but CXCR4 levels elevated at different time points. Surprisingly in PBMCs, no effect of HIV-1 infection on CXCR4 up to 4 hpi and at later time point i.e. at 6 hpi all receptor levels were decreased. Collectively, our results show at early hours of post infection there is less effect of viral factors on the transcriptional regulation of cell surface receptors. The modulations appeared at early hours may be due to the effect of host intrinsic antiviral factors or maybe the involvement of other cell signaling pathways which regulates transcription of these markers. Our data revealed some intra cellular signaling or may be due to the host intrinsic antiviral factors regulates cell surface receptors transcription up to 4 hpi. But at later stages accessory proteins of HIV related from virions might disrupt cellular functions of the infected T-cell even before the time of peak intracellular viral protein and virion production.
Conclusion
Our results support the hypothesis that surface markers of host cells alter very early after the HIV infection may be due to the cell signaling cascades activated during infection. This effects docent involves viral loaded accessory proteins. Over expression of these markers at early hours of post infection instead of decreased levels supports our conclusion. After 4 hpi, the decreased transcription of markers along with constant decrease of gp120 explains the viral loaded accessory proteins role in it. And further analysis of these markers at protein level can help us to understand host cell expression pattern after infection, which can lead to identification of novel surface markers.
Acknowledgement
PSD gratefully acknowledges UGC Dr. Kothari Post-Doctoral Research Fellowships from the University Grants Commission, Govt. of India and SLB acknowledges DST Research Associate fellowship. We thank Prof. Anand K. Kondapi, University of Hyderabad for extending laboratory facilities for these studies and funds from the DST are acknowledged.
References
- Février M, Dorgham K, Rebollo A (2011) CD4+ T Cell depletion in human immunodeficiency virus (HIV) infection: Role of apoptosis. Viruses 5: 586-612.
- Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, et al. (2010) Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell 37: 656-67.
- Merk A, Subramaniam S (2013) HIV-1 envelope glycoprotein structure. Curr Opin Struct Biol 10: 268-76.
- Roitburd-Berman A, Dela G, Kaplan G, Lewis GK, Gershoni JM (2013) Allosteric induction of the CD4-bound conformation of HIV-1 Gp120. Retrovirology 10: 147.
- Lin G, Baribaud F, Romano J, Doms RW, Hoxie JA (2003) Identification of gp120 binding sites on CXCR4 by using CD4-independent human immunodeficiency virus type 2 Env proteins. J Virol 77: 931-42.
- Geiss GK, Bumgarner RE, An MC, Agy MB, Upchurch D, et al. (2000) Large-Scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology 266: 8-16.
- Ringrose JH, Jeeninga RE, Berkhout B, Speijer D (2008) Proteomic studies reveal coordinated changes in T-cell expression patterns upon infection with human immunodeficiency virus type 1. J Virol 82: 4320-30.
- Peraire J, Miró O, Saumoy M, Domingo P, Pedrol E, et al. (2007) HIV-1-infected long-term non-progressors have milder mitochondrial impairment and lower mitochondrially-driven apoptosis in peripheral blood mononuclear cells than typical progressors. Curr HIV Res 5: 467-73.
- Breen EC, McDonald M, Fan J, Boscardin J, Fahey JL (2000) Cytokine gene expression occurs more rapidly in stimulated peripheral blood mononuclear cells from human immunodeficiency virus-infected persons. Clin Vaccine Immunol 7: 5769-773.
- Ali A, Al-Jabri (2003) How does HIV-1 infect a susceptible human cell? Current thinking. J Sci Res Med Sci 5: 31-44.
- Okoye AA, Picker LJ (2013) CD4+ T cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev 254: 54-64.
- Signoret N, Pelchen-Matthews A, Mack M, Proudfoot AE, Marsh MJ (2000) Endocytosis and recycling of the HIV coreceptor Ccr5. Cell Biol 151: 1281-94.
- Shioda T, Nakayama EE, Tanaka Y, Xin X, Liu H, et al. (2001) Naturally occurring deletional mutation in the C-Terminal cytoplasmic tail of CCR5 affects surface trafficking of CCR5A. J Virol 75: 3462-8.
- Cortés MJ, Wong-Staal F, Lama J (2002) Cell surface CD4 interferes with the infectivity of HIV-1 particles released from T cell. J Biol Chem 277: 1770-9.
- Levesque K, Finzi A, Binette J, Cohen A (2004) Role of CD4 receptor down-regulation during HIV-1 infection, Curr HIV Res 2: 51-94.
- Tavares LA, da Silva EM, da Silva-Januário ME, Januário YC, de Cavalho JV, et al. (2017) CD4 downregulation by the HIV-1 protein Nef reveals distinct roles for the γ1 and γ2 subunits of the AP-1 complex in protein trafficking. J Cell Sci 30: 429-43.
- Hatse S, Princen K, Bridger G, De Clercq E, Schols D (2002) Chemokine receptor inhibition byAMD3100 is strictly confined to CXCR4. FEBS Letters 527: 255-62.
- Fernandis AZ, Cherla RP, Chernock RD, Ganju RK (2002) CXCR4/CCR5 Down-modulation and chemotaxis are regulated by the proteasome pathway. J Biol Chem 277: 18111-7.
- Klase Z, Jeang KT (2013) Reciprocal functional pseudotyping of HIV-1 and HTLV-1 viral genomes by the heterologous counterpart envelope proteins. Virology 443: 106-12.
- Holmen SL, Federspiel MJ (2000) Selection of a subgroup A avian leukosis virus [ALV(A)] envelope resistant to soluble ALV(A) surface glycoprotein. Virology 273: 364-73.
- Min F, Man-man D, Ming L, Wei-sheng C (2017) Establishment of an avian leukosis virus subgroup A-resistant cell line. J Integr Agric 16: 930-936.
- Levesque K, Finzi A, Binette J, Cohen EA (2004) Role of CD4 receptor down-regulation during HIV-1 infection. Curr HIV Res 2: 51-9.
- Lindwasser OW, Chaudhuri R, Bonifacino JS (2007) Mechanisms of CD4 downregulation by the Nef and Vpu proteins of primate immunodeficiency viruses. Curr Mol Med 7: 171-84.
- Chenine AL, Sattentau Q, Moulard M (2000) Selective HIV-1-induced downmodulation of CD4 and coreceptor. Arch Virol3: 455-71.
- Valente ST, Chanel C, Dumonceaux J, Olivier R, Marullo S, et al. (2001) CXCR4 is down-regulated in cells infected with theCD4-independent X4 human immunodeficiency virus type 1isolate m7NDK. J Virol 75: 439-47.
- Choi B, Gatti PJ, Fermin CD, Vigh S, Haislip AM, et al. (2008) Down-regulation of cell surface CXCR4 by HIV-1. Virology 5: 6.
- Lapham CK, Romantseva T, Petricoin E, King LR, Manischewitz J, et al. (2002) CXCR4 heterogeneity in primary cells: possible role of ubiquitination. J Leukoc Biol 72: 1206-14.
- Garcia-Perez J1, Rueda P, Staropoli I, Kellenberger E, Alcami J, et al. (2011) New Insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J Biol Chem 286: 4978-90.
- Berger EA, Alkhatib G (2007) HIV gp120 interactions with coreceptors: insights from studies with CCR5-based peptides. Eur J Med Res 12: 403-7.
- Barmania F, Pepper MS (2013) C-C chemokine receptor type five (CCR5): An emerging target for the control of HIV infection. Appl Transl Genom 2: 3-16.
- Cormier EG, Persuh M, Thompson DA, Lin SW, Sakmar TP (2000) Specific interaction of CCR5 amino-terminal domain peptides containing sulfotyrosines with HIV-1 envelope glycoprotein gp120. Proc Natl Acad Sci U S A 97: 5762-7.