Keywords
Malformations; Hypoplasia; Fetus; Syndrome; Proliferation; Sonographic
Abbreviations
EFTUD2: Elongation Factor Tugtp Binding Domain containing 2; ESCO2: Establishment of Cohesion1, S, Cerevisiae, Homolog of 2 Ecol1, S Cerevisiae, Homolog of 2; MC3: Melanocortin 3 Receptor Gene; NIPBL: Nipped-B-like Protein (NIPBL), also known as Delangin or SCC2 Homolog; POLR1D: Polymerase (RNA) I Polypeptide D; POLR1C: Polymerase (RNA) I Subunit C; RECQL4: RecQ Protein-like 4; RBM8A: RNA Binding Motif Protein 8A; SMC1A: Structural Maintenance of Chromosomes 1A; TCOF1: “Treacher Collins-Franceschetti Syndrome 1 Gene Provides Instructions for Making a Protein Called Treacle; TBX5: T-Box Transcriptipon Factor
Introduction
Among the heterogeneous group of acrofacial dysostoses (AFD1) involving craniofacial and upper limb abnormalities, the incidence of its prototype, Nager syndrome (NS)(OMIM# #154400), has been reported to be 3:1,000,000 [1]. NS, characterized by mandibulofacial dysostosis with preaxial limb malformations, consists of downslanting of palpebral fissures, malar and mandibular hypoplasia, micrognathia, low-set and dysplastic ears, preauricular tags, cleft palate, bilateral radial agenesis, club hands and hypoplastic or absent thumbs. The syndrome, though there are even conflicts of its being a syndrome, as Opitz suggested that the Nager syndrome represented an 'anomaly' rather than a syndrome because of its causal heterogenity [2], was recognized as a specific entity by Nager and de Reynier [3]. However, Gobbel et al., cites the Meckel specimen from 19th Century, in the Meckel anatomic collection at the University of Halle, Berlin, as the earliest known fetus with Nager AFD [4]. Radiological and CT examination demonstrated his mandibular dysplasia, dysgenesis of the ear capsule, bilateral radial agenesis with laterally convex diaphysis of the ulnae, left sided aplasia and right sided hypoplasia of the thumbs, bilateral club hands, varus, adduction and supination of the right foot together with outward rotation of the face of the left tibia. Among approximately 100 patients with NS described in the literature [5], in none of them has a karyotype anomaly or gene defects been reported [6]. Though the etiology of NS remains in question, a wide range of variability suggests genetic heterogeneity [7] Both autosomal recessive and dominant inheritance with reduced penetrance have been suggested, however, most cases have been sporadic with no reports of affected parents [8]. Recent studies [7] used exome sequencing as a discovery tool and found mutations in spliceosomal factor 3 B4(SF3B4), a component of the U2 premessenger ribonucleic acid (RNA) splicesomal complex causing NS. Proper splicing of premessenger RNA is required for protein synthesis and therefore fundamental for cellular function. SF3B4 gene mutation on mutation analysis of coding exons has been postulated to produce a defective balance between protein isoforms leading to functional consequences, including defective regulation of proliferation and differentiation that may be associated with NS. However, this may not be the case for phenocopies or in the presence of consanguinity [9]. The aim of this paper is to report the first trimester sonographic features, postmortem findings and the negative result of the array based technological assessment of the recently discovered mutations in SF3B4 protein genes that constitute of a novel class of genomic lesions for NS.
Clinical Report
A 23 year old primigravid woman, first cousin with her husband from both mother and father, evaluated for vaginal bleeding at 6 weeks of gestation.
Ultrasonographic examination showed an embryo with cardiac activity compatible with her last menstrual period, and a closed cervical canal. She was not exposed to drugs, radiation or any known teratogen. Sonographic scan at 11 weeks of gestation revealed a nuchal thickness of 3.0 mm with abnormal facial profile (Figure 1).
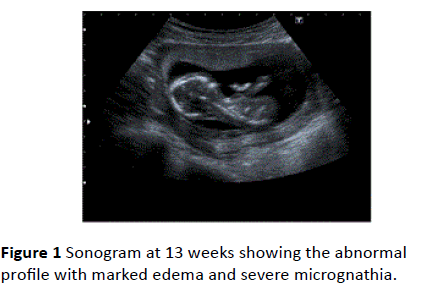
Figure 1 Sonogram at 13 weeks showing the abnormal profile with marked edema and severe micrognathia.
Biparietal diameter (BPD) and head circumference (HC) measurements were below the 10th percentile at 13 weeks. Severe micrognathia and malar hypoplasia (Figure 1) and the club hand deformity together with hypoplastic/absent thumbs and fixed elbows and wrists were also present (Figure 2).
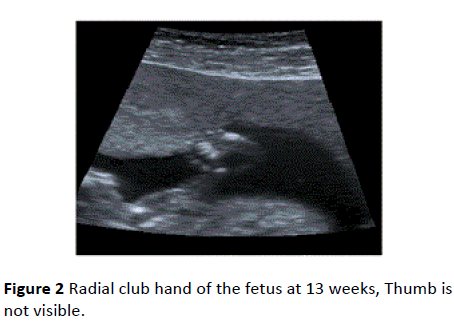
Figure 2 Radial club hand of the fetus at 13 weeks, Thumb is not visible.
Biochemical screening results at 11 weeks were: Associated Plasma Protein A (PAPP-A) of 1.32 mIU/ml (0.59 MOM) and a free β human chorionic gonadotropin (hCG) of 15.1 ng/ml (0.30 MOM), (Immulite PAPP-A, free βhCG control module, PILPCCM-45 and PILFBCM-52, Siemens Medical Solutions CA, USA) with a risk of 1/720 in combined test for trisomy 21 and an increased risk for trisomy 13/18+NT (1/356) (The Prisca Typolog software GmBH, Germany). Chorion villus sampling (CVS) was performed. Fetal karyotype analysis revealed 46 XY karyotype. Vaginal bleeding persisted after CVS. Due to the presence of multiple malformations, parents were counselled and the pregnancy was terminated at 15 weeks.
A 30 gram/10 cm male infant was delivered after vaginal misoprostol induction. Prenatal sonographic findings:
• Small atypical head
• Underdevelopment of the jaw area and cheek
• Bilateral radial aplasia
• Bilateral club hands
• Absence/hypoplasia of thumbs were evident at postmortem examination (Figures 3 and 4).
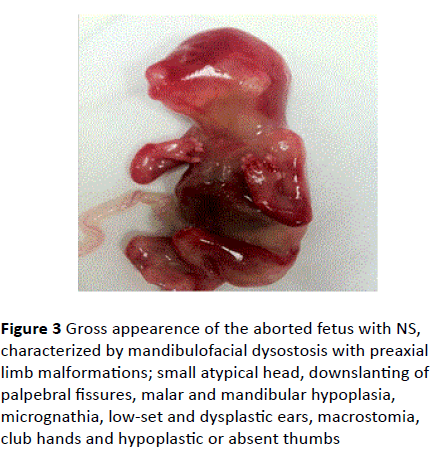
Figure 3 Gross appearence of the aborted fetus with NS, characterized by mandibulofacial dysostosis with preaxial limb malformations; small atypical head, downslanting of palpebral fissures, malar and mandibular hypoplasia, micrognathia, low-set and dysplastic ears, macrostomia, club hands and hypoplastic or absent thumbs
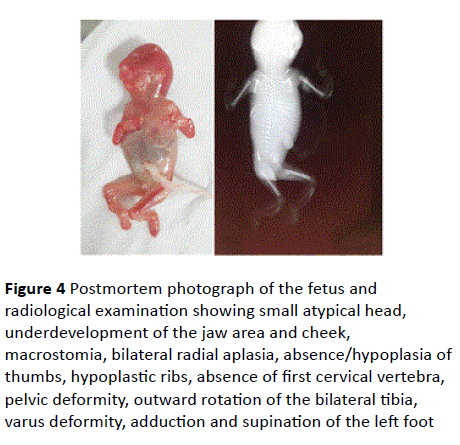
Figure 4 Postmortem photograph of the fetus and radiological examination showing small atypical head, underdevelopment of the jaw area and cheek, macrostomia, bilateral radial aplasia, absence/hypoplasia of
thumbs, hypoplastic ribs, absence of first cervical vertebra, pelvic deformity, outward rotation of the bilateral tibia, varus deformity, adduction and supination of the left foot
Downslanted palpebral fissures, dislocated ears, macrostomia, cleft palate, hypoplastic ribs, absence of first cervical vertebra, pelvic deformity, outward rotation of the bilateral tibia, varus deformity, adduction and supination of the left foot were noted at postmortem examination and Xrays (Figures 3-5).
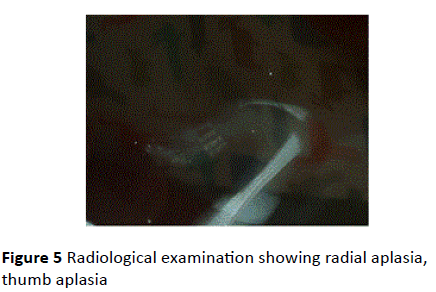
Figure 5 Radiological examination showing radial aplasia, thumb aplasia
Genomic DNA was extracted from aborted fetus, CVS material and peripheral blood from both parents. Fetal DNA was analyzed for SF3B4 mutations by Sanger sequencing. The entire SF3B4 coding region and splicing junctions were amplified in 7 PCR fragments. The following primer pairs were used (Table 1).
| Forward |
Reverse |
| 5'- AAGTGGAAGTCGTGCTGAGG-3' |
5'- CA TGAAGATGGAACCCAACC-3' (Exon 1) |
| 5'-TTCCTTCCTTCCCGTTACAC-3' |
5'-TTGTGAATACTGCTGGGACC-3' (Exon 2) |
| 5'-TGAATTTTACCCCATTTTCAGTC-3' |
5'- GGACGGTTACAGAGGTACTGC-3'(Exon 3) |
| 5'-TGGGGTCATCTTACAAACCC-3' |
5'-TTTCTTCTTCCTCCTGACCC-3' (Exon 3) |
| 5'-AGGCCAGATCAGGACAGG-3' |
5'-CTGTTGAGGAACAAAGGGCA-3' (Exon 4) |
| 5'-GCCAGCCTTATTTTCTATACCC-3' |
5'-TAGTAAGGGCACGGGACAAG-3 (Exon 5) |
| ' 5'-TCTAGCCACCTCCCTCATCT-3' |
5'- GGATTAGTACCTTTGCCCCA-3' (Exon 6) |
Table 1: primer pairs
Primers were designed using the NCBI/Primer-BLAST software [10] and synthesized by Metabion (International AG, Martinsried, Deuschland). PCR reactions were carried out under standard conditions. Direct sequencing was performed using a ABI PRISM BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies, Foster City, Calif., USA) and the standard protocol. Electropherograms were compared with the SF3B4 mRNA reference sequence 7 (NM_005850).
Mutation analysis of coding exons of SF3B4 did not identify any pathogenic mutations 8 months after the termination of patient's first pregnancy, a 6 week spontaneous abortion occurred.
Discussion
Congenital anomalies are common in human reproduction and have a prevalence of 5%. First and second trimester imaging for all pregnancies is a well-recognized “standard of care”. Sonographic imaging has been reported to identify the majority of major “structural” anomalies that account for approximately 70% of the congenital burden. When defects of the craniofacial and limb development are detected during sonographic examination, a karyotype analysis, either by cell free DNA or invasive tests may be used to exclude Trisomy 18, Trisomy 13, Ring D chromosome abnormalities [11-13]. After excluding karyotype aberrations, differentiation from other entities that may resemble NS should be carried out clinically (Table 2).
| |
Typical Clinical Findings |
Genetic Changes |
| Trisomy 18 |
Multiple malformations, clenched hand, craniofacial defects, growth restriction, mental retardation, abnormal hands and feet [11] |
Numerical chromosomal anomaly |
| Trisomy 13 |
Multiple malformations, holoprosencephaly, facial defects [12] |
Numerical chromosomal anomaly |
| Ring D chromosome abnormalities |
Mental retardation, facial dysmophic features, short stature [13] |
Deletions occurring at both ends of the same chromosome, the ends may come together to form a ring chromosome |
| Nager Syndrome OMIM# #154400 |
Mandibulofacial dysostosis with preaxial limb malformations downslanting of palpebral fissures, malar and mandibular hypoplasia, micrognathia, low-set and dysplastic ears, preauricular tags, cleft palate, bilateral radial agenesis, club hands, hypoplastic/absent thumbs |
Splicesomal Factor 3B4 deficiency in 60% of cases |
| TAR syndrome (OMIM# 274000) |
Thrombocytopenia, Absent radius, Thumb always present |
RBM8A gene on chromosome 1 |
| Miller (Genée-Weidemann or postaxial acrofacial dysostosis) syndrome (OMIM# 263750) |
AFD type II with post-axial malformations |
Dihydroorotate (DHDOD) gene located at chromosome 16q22. This gene encodes DHDOD enzyme |
| Mandibulofacial dysostosis with microcephaly (MFDM) (OMIM #610536) |
Progressive microcephaly, midface and malar hypoplasia, micrognathia, dysplastic ears, preauricular microtia, skin tags, significant developmental delay, cleft palate |
EFTUD2 gene on chromosome 17q21 |
| Baller Gerold syndrome (OMIM# 218600) |
Abnormally shaped skull, coronal craniosynostosis. Short stature, aplasia /hypoplasia of the radius |
Homozygous or compound heterozygous mutation in the RECQL4 |
| Cornelia de Lange syndrome (OMIM# 122470) |
Facial defects, growth restriction and mental retardation, upper limb anomalies |
Defects in chromosome 5,10,X NIPBL, SMC1A, MC3 genes |
| Fanconi anemia (OMIM# 613390 RAD51C) |
Absent thumbs, radial ray defects, congenital hip dislocation, scoliosis vertebral anomalies, microphtalmia, microcephaly, conductive deafness developmental delay, high incidence of genital abnormalities |
Chromosomal hyper sensitivity to cross-linking- agents, such as mitomycine butane C / diepoxy resulting increase in chromosome breakage in males provides the basis for a diagnostic test |
| Holt±Oram syndrome (OMIM# 142900) |
Upper extremity abnormalities, heart defects |
TBX5 gene |
| Treacher-Collins Franceshetti syndrome (OMIM# 154500) |
Antimongoloid slant of the eyes, coloboma in the lower eye lid, micrognathia, microtia and other deformity of the ears, hypoplastic zygomatic arches, macrostomia, cleft palate |
TCOF1 on chromosome 5q32 POLR1D on chromosome 3q12.2 POLR1C on Chromosome 6 |
| Goldenhar syndrome (Oculo-Auriculo Vertebral Syndrome Hemifacial microsomia Syndrome) (OMIM# 164210) |
Incomplete development of the ear nose, soft palate, lip and mandibular usually on only one side, hemivertebrae vertebral hypoplasia, block vertebral anomalies, coloboma in the upper eye lid |
not genetically linked |
| Pierre-Robin syndrome (OMIM# 261800) |
cleft palate, retrognathia, glossoptosis |
genetic anomalies at chromosomes 2, 11, or 17 |
| Roberts syndrome (OMIM# 268300) |
Severe shortening of limbs, facial anomalies corneal opacities |
ESCO2 gene |
| VATER/ VACTERL associations (OMIM# 192300) |
Vertebral defects, anal atresia, tracheo-esophageal fistula, radial/renal dysplasias |
|
Table 2 Major differential diagnoses of Acrofacial Dysostosis
Clinically, the presence of anterior upper-limb defects and the typical lack of lower-limb involvement distinguishes Nager syndrome from TAR syndrome with the presence of both thumbs in the latter. Preaxial upper limb defects point to the AFD NS, whereas postaxial limb defects point to the Gene´e- Wiedemann or Miller syndrome. There is no limb defect in Tracher Collins- Franceshetti syndrome. In Holt Oram and Fanconi syndrome, there is no mandibular and malar hypoplasia. Diagnostically relevant are: Goldenhar syndrome, Pierre-Robin syndrome and Vater associations with the absence of radial and thumb aplasia in all. The diagnosis of MFDM was also suspected in the present case. As the phenotypic overlap between MFDM and NS is substantial, gene analysis and exome sequencing may be conducted (Table 2), however, to our knowledge, SF3B4 gene mutation in NS has not been reported from 8 consanguineous parents. EFTUD2 analysis has been reported to be considered in all cases with clinical suspicion of NS and no mutation in SF3B4.
As a conclusion, out of approximately 100 cases that have been reported so far, this is the first paper presenting the first trimester sonographic findings of NS. The diagnosis of NS may be feasible at first trimester by being aware of the syndrome and be considered if concomittant craniofacial and limb defects are detected during sonographic examination; however no mutation has been identified in SF3B4. As exome sequencing is only able to identify those variants found in the coding region of genes which affect protein function, structural and non-coding variants associated with the disease can not be identified using this method. Whole genome sequencing may be useful in the present case with no mutation in SF3B4. There remains 99% of the human genome that is not covered using exome sequencing. Whole genome sequencing may become a standard approach and allow a deeper understanding of genetic variation found in Nager syndrome and its phenocopies, but presently, this technique is still not practical due to the high costs and time associated with sequencing large numbers of genomes.
References
- Halonen K, Hukki J, Arte S, Hurmerinta K (2006) Craniofacial structures and dental development in three patients with Nager syndrome. J Craniofac Surg 17: 1180-1187.
- Opitz, JM (1987) Nager "syndrome" versus "anomaly": and its nosology with the postaxial acrofacial dysostosis syndrome of Genee and Wiedemann. Am J Med Genet 7: 959-63.
- Nager FR, Reyner JP (1948) Das Gehörorgan bei den angeborenen KopfMisbuldungen. Pract Oto Rhinolaryngol 10: 28.
- Gobbel L, Schultka R, Klunker R, Stock K, Wand D, et al. (2007) Acrofacial Dysostosis (AFD) with preaxial limb hypoplasia (Nager AFD) and club foot diagnosed in a fetus from 1812 in the Anatomical Collections at the University of Halle, Germany. Am J Med Genet 137A: 263 - 8.
- Paladini D, Tartaglione A, Lamberti A, Lapadula C, Martinelli P (2003) Prenatal ultrasound diagnosis of Nager syndrome. Ultrasound Obstet Gynecol 21: 195-197.
- Schlieve T, Almusa M, Miloro M, Kolokythas A (2012) Temporomandibular joint replacement for ankylosis correction in Nager syndrome: case report and review of the literature. J Oral Maxillofac Surg 70: 616-625.
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, et al. (2011) Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745-755.
- Bernier FP, Caluseriu O, Ng S, Schwartzentruber J, Buckingham KJ, et al. (2012) Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet 90: 925-933.
- Czeschik JC, Voigt C, Alanay Y, Albrecht B, Avci S, et al. (2013) Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum Genet 132: 885-98.
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, et al. (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13: 134.
- Lin HY, Lin SP, Chen YJ, Hung HY, Kao HA, et al. (2006) Clinical characteristics and survival of trisomy 18 in a medical center in Taipei, 1988-2004. Am J Med Genet A 140: 945-951.
- Lin HY, Lin SP, Chen YJ, Hsu CH, Kao HA, et al. (2007) Clinical characteristics and survival of trisomy 13 in a medical center in Taiwan, 1985-2004. Pediatr Int 49: 380-386.
- Cunningham FG, Leveno KJ, Bloom SI, Dashe JS, Hoffman BI, et al. (2014) Genetics-in Williams Obstetrics (24thedn) Mc GrawHill Education, Newyork.