Keywords
Acyclovir sodium, Sustain release, Solid lipid micro particles
Introduction
Acyclovir is an antiviral drug which slows the spread and growth of herpes virus. It is also used to treat genital herpes, cold sores, shingles and chicken pox. Acyclovir has a low bioavailability of 20-30% and excreted in unchanged in urine both by filtration and secretion. Hence it is prescribed 3-4 times a day which has negative effect on patient compliance [1]. Acyclovir may also cause side effects like bleeding, purple or red pinpoint spots under skin, no urinating , painful or difficult urination, swelling in feet or ankles, feeling tired or short of breath. In order to enhance the efficacy and to improve safety formulation of solid lipid micro particles is carried out. Solid lipid micro particles are having size between in the range of 1-1000 μm with drug being dissolved, entrapped and encapsulated in micro particle matrix [2].
The drug entrapment got upto 80% which are easily compatible with living systems since SLMs system based of biomaterials. In SLMs drug is protected from degradation as it is sealed in the biomaterial matrix. The leaching of the drug is passive mechanism and is independent of concentration of drug. As the micro particle continues get dissolved leading to slow sustained release of the drug in the stomach. The suitability of solid lipid micro particles for sustained release has been established by several authors [3-5]. SLMs can be prepared by Spray-drying, Spray congealing, O/W melt dispersion technique, Double-emulsion solvent evaporation, solvent evaporation, High pressure homogenization, w/o melt dispersion technique [6]. The improvement in patient can be dosing schedule can be reduced one or two times. In this paper SLMs of Acyclovir sodium of different concentrations of bio lipids are prepared using stearic acid by o/w melt dispersion techniques. The various formulations are evaluated for suitability of formulation of sustained released preparation [7].
Materials and Methods
Material
All the reagents and chemical used are of analytical grade doubly distilled water was used in making preparation in this study. Acyclovir sodium (Helix Pharma Pondicherry, India), Stearic acid (Lipidchem Johor, Malaysia), Tween 80 (Thomas baker Mumbai, India), Dimethyl sulfoxide (SD fine chemicals, Mumbai, India).
Preparation of Solid Lipid Micro Particles containing Acyclovir Sodium
Two different methods were used for preparation of solid lipid micro particles with Dimethyl sulfoxide. In method 1 SLMs were prepared in o/w melt preparation technique. Stearic acid was melted on a water bath with a temperature limit of 72°C. The drug particles grounded to the fine size was dispersed with molten stearic acid aqueous phase consisting of water and tween 80 at 72°C was the molten phase was slowly added to the aqueous phase by steering and the emulsification was assisted by homogenizer for 15 minutes8. The dispersion rapidly cools down at 20°C by immersing the solution in ice bath. The formed micro particles were separated by the filtration by this formulation F1, F2, F3 and F4 were prepared. In another method formulation F7 and F8 were prepared by same method. However the Dimethyl sulfoxide is used instead of water and tween 80 to dissolve finally grounded acyclovir sodium (Table 1).
| Formulation |
Drug (gm) |
Lipid stearic acid (gm) |
Tween80 (ml) |
Dimethyl Sulfoxide (ml) |
Water q.s (ml) |
| F1 |
0.2 |
1 |
0.1 |
- |
100 |
| F2 |
0.2 |
2 |
0.1 |
- |
100 |
| F3 |
0.2 |
3 |
0.1 |
- |
100 |
| F4 |
0.2 |
4 |
0.1 |
- |
100 |
| F5 |
0.2 |
3 |
0.1 |
2 |
100 |
| F6 |
0.2 |
3 |
0.1 |
3 |
100 |
| F7 |
0.2 |
3 |
0.1 |
4 |
100 |
| F8 |
0.2 |
1 |
0.1 |
3 |
100 |
Table 1: Formulation details [9]
Characterization of SLMs of Acyclovir Sodium: The characterization of Acyclovir was carried out using FT-IR spectrum and Scanning Electron Microscopy [10].
FT-IR spectrum: The KBr disc technique was employed for preparation of sample. Pellet of 1 mg Acyclovir sodium and 100 mg dried spectroscopic grade KBr was prepared in a die with the application of pressure and pellet was analyzed using FT-IR Spectrophotometer.
Scanning electron microscopy: Scanning electron microscopy (SEM) was used to verify uniformity of particle shape and size. The samples were previously fixed on a brass stub using double-sided adhesive tape and were then made electrically conductive by coating with a thin layer of gold and palladium alloy (180- 200Å) using a fine coat ion sputter (SUK PHY) (JEOL, fine coat ion sputter JFC-1100). The surface morphology of the sample was observed under a scanning electron microscope JSM-6400 (Joel, Japan) operated at 10kev pulse at different resolutions [11].
Evaluation of SLMs
Particle size: The particle sizes of the microparticles were determined by using optical microscopy method. Approximately 100 microparticles were counted for particle size analysis by using calibrated optical microscope [12,13].
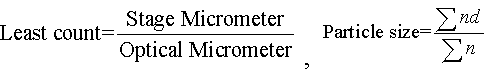
Here, Σn = no. of particles
Σnd = average diameter
Percentage (%) yield and drug loading capacity: The prepared microparticles were collected and weighed. The measured weight was divided by the total weight of all the excipients and drug. To calculate the drug loading capacity, weighed 10 mg drug was added to 10 ml of phosphate buffer (pH 6.8) to facilitate the microparticles to get dissolved and heated at 70°C,then the resultant solution centrifuged at 8000 rpm for 10 min. Samples were measured at an absorbance of 252 nm in double beam U.V Spectrophotometer. Percentage yield and drug loading capacity of microparticles were determined by the following equations:
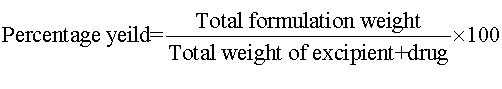
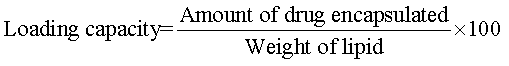
Angle of repose: Angle of repose of different formulations was measured according to fixed funnel standing method.
Here, θ=angle of repose, r=radius, h=height
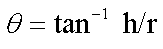
Bulk density and tapped density: Bulk and tapped densities were measured by using 10 ml of graduated cylinder. The sample was poured in cylinder, tapped mechanically for 100 times, then tapped volume was noted down to calculate bulk density and tapped density. Each experiment for micromeritic properties was performed in triplicate manner, by using following equation.
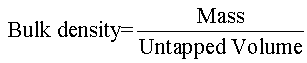
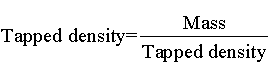
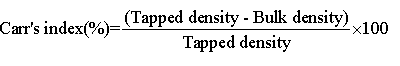
Hausner’s ratio and Carr’s index: Hausner’s ratio of microparticles was determined by comparing the tapped density to the bulk density using the equation. Carr’s index value of microparticles was computed according to the following equation:
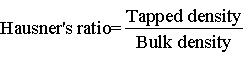
In Vitro Dissolution Studies
In vitro drug release study was carried out using USP Dissolution apparatus (Type II). The formulations equivalent to weight containing 200 mg acyclovir sodium were immersed in dissolution medium. Phosphate buffer (pH 6.8) (900 ml) was used as the dissolution medium and maintained at 37 ± 0.5°C at a rotation speed of 50 rpm. Samples were withdrawn and the same amount was replaced using the same dissolution medium. The amount of the drug was quantified using double beam U.V spectrophotometer at 252 nm [14].
Drug Release Kinetic Studies
Raw data obtained from in vitro release studies was analyzed,and it was fitted to different equations and kinetics model to calculate the percent drug release and release kinetics of acyclovir sodium from microparticles. The kinetic models used were zero-order equation, first-order, higuchi’s model and korsmeyerpeppas equation15.
Results
Characterization and Evaluation of SLMs
FT-IR spectrum: Peaks were evident at 3572.41 cm-1 (O-H stretching), 3342.62 (O-H stretching), 2915.43 (aliphatic C-H stretching), 2873.38 (aliphatic C-H stretching symmetric), 1632.41 (0-H deformation), 1459.09 (aliphatic C-H deformation), 1278.57 (C-O stretching) for Acyclovir sodium showed in Figure 1. So presence of these characteristic peaks of Acyclovir sodium in physical mixture reveals that the drug remains intact in formulation F5 and there is no interaction between drug and stearic acid used Figure 2.
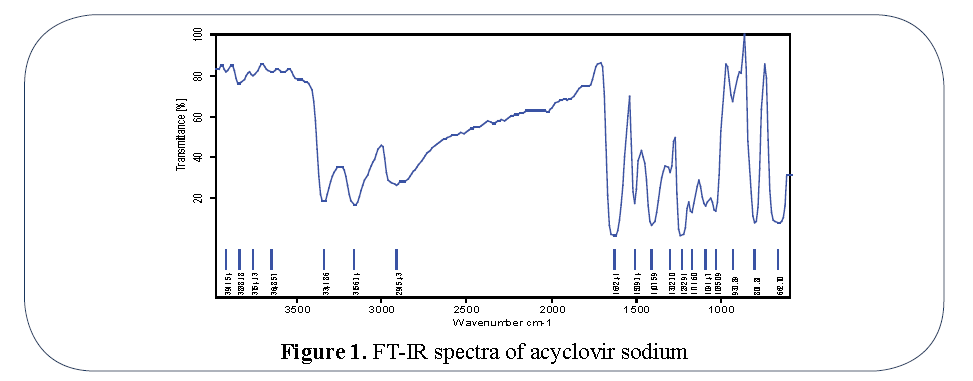
Figure 1: FT-IR spectra of acyclovir sodium
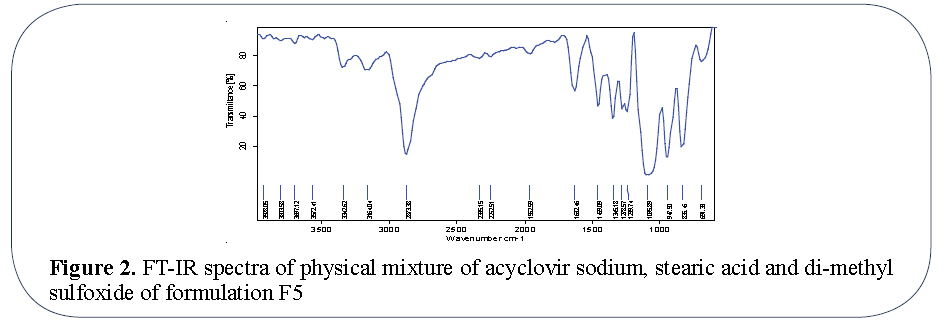
Figure 2: FT-IR spectra of physical mixture of acyclovir sodium, stearic acid and di-methyl sulfoxide of formulation F5
Scanning electron microscopy: Scanning electron microscopy was performed for lipid microparticles of formulation F5 to obtain more information on the particle size and morphology. The photos of lipid microparticles shows that the formulated acyclovir sodium microparticles were of spherical shape, which were shown in Figure 3. Moreover the microparticles were observed with smooth surface, which may contribute to its release of the drug in a sustained manner compared to the microparticles having rough surface.

Figure 3: SEM image of acyclovir sodium SLMs of formulation
Evaluation of SLMs
Particle size, percentage yield and drug loading capacity: The reason behind selecting solid lipid microparticles was their ability to have better particle size (7 μm), percentage yield and to better drug entrapment. Drug loading capacity is considered as an important parameter as improper entrapment leads to the initial burst release of the drug, which hinders its sustained release property. Acyclovir sodium solid lipid microparticles of formulation F5 has shown 100% drug loading capacity as depicted in Table 2.
| Formulation code |
Particle size (µm) |
Percentage yield |
Drug loading capacity |
| F1 |
4.96 |
39.7 |
9.6 |
| F2 |
4.92 |
54.6 |
14.4 |
| F3 |
4.61 |
76.9 |
57.3 |
| F4 |
4.18 |
83.8 |
11.4 |
| F5 |
7.67 |
97.8 |
100 |
| F6 |
6.20 |
87.5 |
94.6 |
| F7 |
6.72 |
78.1 |
50 |
| F8 |
4.36 |
26.7 |
1.2 |
Table 2: Particle size, percentage yield and drug loading capacity
Bulk density and tapped density: The bulk density and tapped density of the formulations were in range of 0.18 to 0.31 and 0.25 to 0.39 respectively. Formulation F5 was having Bulk density=0.31 and Tapped density=0.39 as depicted in Table 3.
| Formulation code |
Bulk density |
Tapped density |
Hausner’s ratio |
Carr’s index |
Angle of repose |
| F1 |
0.29 |
0.39 |
1.34 |
25.6 |
33 |
| F2 |
0.26 |
0.33 |
1.26 |
21.2 |
37 |
| F3 |
0.28 |
0.36 |
1.28 |
22.2 |
36 |
| F4 |
0.29 |
0.36 |
1.24 |
19.4 |
32 |
| F5 |
0.31 |
0.39 |
1.25 |
20.5 |
30 |
| F6 |
0.28 |
0.35 |
1.25 |
20 |
32 |
| F7 |
0.18 |
0.25 |
1.38 |
28 |
32 |
| F8 |
0.27 |
0.29 |
1.07 |
6.89 |
33 |
Table 3: Bulk density, tapped density, hausner’s ratio, carr’s index and angle of repose
Hausner’s ratio and Carr’s index: Hausner’ ratio were found to be in the range 1.07 to 1.38. Carr’s Index was in the range of 6.89 to 28. Formulation F5 was having Hausner’s ration 1.25 and Carr’s index 20.5 as depicted in Table 3.
Angle of repose: Angle of repose values of formulations F1 to F8 were found in range 30 to 37. The formulation F5 was having angle of repose 30 as depicted in Table 3.
In vitro Drug Release
The percentage drug release from acyclovir sodium with formulation F5 was observed by using USP Dissolution tester (Type II). These were compared with the pure acyclovir sodium drug as shown in Figure 4. The in vitro drug release showed that the solid lipid microparticles containing acyclovir sodium have sustained the drug release. Formulation F5 showed better sustained release of drug. In vitro release of formulation F5 was 75% after 17hrs shown in Table 4.
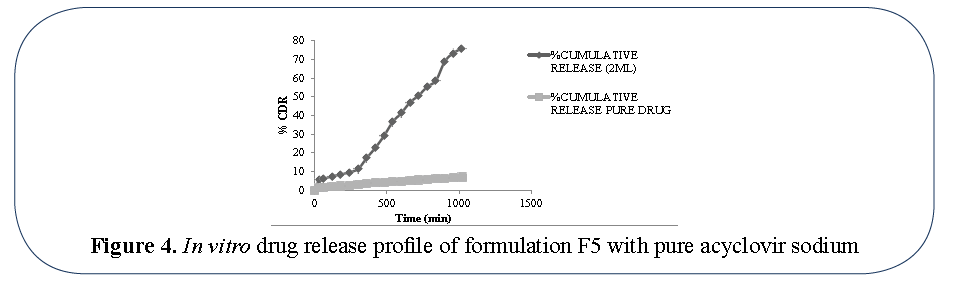
Figure 4: In vitro drug release profile of formulation F5 with pure acyclovir sodium
| Time (min) |
Log time |
S.R time |
%cumulative release |
Log % cumulative release |
% cumulative remaining |
Log % cumulative remaining |
| 0 |
0 |
0 |
0 |
0 |
100 |
2 |
| 30 |
1.477121255 |
5.477225575 |
5.497826 |
0.740190991 |
94.502174 |
1.975441799 |
| 60 |
1.77815125 |
7.745966692 |
6.032196 |
0.780475444 |
93.967804 |
1.972979078 |
| 120 |
2.079181246 |
10.95445115 |
7.525848 |
0.876555443 |
92.474152 |
1.966020357 |
| 180 |
2.255272505 |
13.41640786 |
8.443978 |
0.926547093 |
91.556022 |
1.961686915 |
| 240 |
2.380211242 |
15.49193338 |
9.676163 |
0.985703176 |
90.323837 |
1.955802378 |
| 300 |
2.477121255 |
17.32050808 |
11.38905 |
1.0564875 |
88.61095 |
1.947487393 |
| 360 |
2.556302501 |
18.97366596 |
17.27123 |
1.237323268 |
82.72877 |
1.917656567 |
| 420 |
2.62324929 |
20.49390153 |
22.76862 |
1.357336709 |
77.23138 |
1.887793795 |
| 480 |
2.681241237 |
21.9089023 |
29.15253 |
1.464676251 |
70.84747 |
1.850324346 |
| 540 |
2.73239376 |
23.23790008 |
36.52177 |
1.562551817 |
63.47823 |
1.802624809 |
| 600 |
2.77815125 |
24.49489743 |
41.35568 |
1.616535166 |
58.64432 |
1.768225955 |
| 660 |
2.819543936 |
25.69046516 |
46.97753 |
1.671890179 |
53.02247 |
1.724459955 |
| 720 |
2.857332496 |
26.83281573 |
50.74688 |
1.705409346 |
49.25312 |
1.692433747 |
| 780 |
2.892094603 |
27.92848009 |
55.40079 |
1.743515958 |
44.59921 |
1.649327166 |
| 840 |
2.924279286 |
28.98275349 |
58.78808 |
1.769289277 |
41.21192 |
1.615022848 |
| 900 |
2.954242509 |
30 |
68.7334 |
1.837167827 |
31.2666 |
1.495080658 |
| 960 |
2.982271233 |
30.98386677 |
73.4071 |
1.865738067 |
26.5929 |
1.4247657 |
| 1020 |
3.008600172 |
31.93743885 |
75.8359 |
1.879874845 |
24.1641 |
1.383170624 |
Table 4: Drug release of acyclovir sodium solid lipid microparticles for F5 formulation
Discussion
Acyclovir sodium formulations with DMSO and stearic acid and without DMSO has produced solid lipid micro particles with desirable flow properties and formulation F5 was found to be most suitable. The FT-IR studies indicated no chemical interactions between Acyclovir and stearic acid. The Tapped density (0.31 and 0.39), Carr’s and Hausner’s ratio (20.5 and 1.25), Angle of repose (30) is suitable for punching and capsulation of the Acyclovir sodium solid lipid micro particles. In vitro dissolution studies has established the SLMs release the Acyclovir sodium in a sustained manner of zero Oder release which was statically with high or values. Slow sustained release of micro particles is going to enhance the bioavailability of drug and suitable for the design of sustained released formulation. The sustained release shall be able to do away with dosing schedules with acyclovir sodium. Sustained release preparation would belief improve the patient compliance can be improve the outcomes to herpes infection treatment.
Acknowledgement
The authors are thankful to Dr Sarvesh Malvia Jain, Director, Oniosome Healthcare Pvt. Ltd, Mohali, Punjab for providing the assistance in carrying out the research work.
References
- Gunness P, Aleksa K, Koren G. Acyclovir-induced nephrotoxicity: the role of the acyclovir aldehyde metabolite. Transl Res. 2011;158(5):290-301.
- Antonio JA, Eliana S. Solid lipid nanoparticles as a drug delivery system for peptides and proteins.Adv Drug Deliv Rev. 2007;59(6): 478-90.
- Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs.J Control Release. 2008;129(1):1-10.
- Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery- a review of the state of the art.Eur J Pharm Biopharm. 2000;50(1):161-77.
- Berton A, Piel G, Evrard B. Powdered lipid nano and microparticles: production and applications.Recent Pat Drug DelivFormul. 2011;5:188-200.
- Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system.Eur J Pharm Sci. 2006;29(3-4):278-87.
- Kawakami K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs.Adv Drug Deliv Rev. 2012; 64(6):480-95.
- Almeida H, Amaral MH, LobaoP.Comparative study of sustained-release lipid microparticles and solid dispersions containing ibuprofen.BrazJ Pharm Sci. 2012;48(3):530-36.
- Sanna V, Kirschvink N, Gustin P, et al.Preparation and in vivo toxicity study of solid lipid microparticles as carrier for pulmonary administration. AAPS Pharm Sci Tech. 2004;5:17-23.
- Bhise KS, Mohammed N. Formulation and development of fenofibrateloaded lipospheres system.JDDT. 2013;3(1):1-10.
- Dalpianz A, Mezzena M, Scatturin A, et al.Solid lipid microparticles for the stability enhancement of the polar drug N6-cyclopentyladenosine. Int J Pharm. 2008;355(1-2):81-6.
- Eradel MS, Gungor S, Ozsoy Y,et al.Preparation and in vitro evaluation of indomethacin loaded solid lipid Microparticles.Act Pharm Sci. 2009;51:203-10.
- Savolainen M, Khoo C, Glad H,et al.Evaluation of controlled release polar lipid Microparticles.Int J Pharm. 2002;244(1-2):151-61.
- Kushwaha A, Prajapati S, Sharma B. Comparative study of acyclovir solid dispersion for bioavailability enhancement. American research of pharmtech research. 2011;1:179-201.
- Chambi HNM, AlvimID,Barrera-Arellano D, et al.Solid lipid microparticles containing water-soluble compounds of different molecular mass: production, characterization and release profile. Food Res Int. 2008;41:229-36.