Keywords
Epigenetics; Histone deacetylases; Inhibitors; Oncology; Inflammation; Neurology; Virology
Abbreviations
HDAC: Histone Deacetylase; HDACi: Histone Deacetylase Inhibitor; HAT: Histone Acetyltransferases; RPD3: Histone deacetylase; SIRT: Sirtuins; NAD: Nicotinamide Adenine Dinucleotide; CAP: Cap Group; CU: Connection Unit; HS: Hydrophobic Spacer; ZBG: Zn-Binding Group; FDA: Federal Drug Administration; CTCL: Cutaneous T Cell Lymphoma; PTCL: Peripheral T-Cell Lymphoma; mTOR: Mechanistic Target of Rapamycin; DNA: Deoxyribonucleic Acid; AML: Acute Myeloid Leukemia; MDS: Myelodysplastic Syndromes; DNMTi: DNA Methyltransferase Inhibitor; VPA: Valproic Acid; LSD1: Lysine (K)-Specific Demethylase 1A; BIM: Proapoptotic Protein; TRIAL: Tumour Necrosis Factor Related ApoptosisInducing Ligand; TSA: Trichostatin A; HosDXR150: Multidrug Resistant Osteosarcoma Cell Line; AMWAP: Activated Microglia / Macrophage WAP Domain Protein; CD44: Cell Surface Glycoprotein; HMGR: 3Hydroxy3Methylglutaryl CoA Reductase; Wee1: Nuclear Kinase; TDP-A: Thailandepsin A; Bax/Bcl2: Proapoptotic Protein; PARP: Poly (ADP-ribose) Polymerase; siRNA: Small Interfering Ribonucleic Acid; APL: Acute Promyelocytic leukemia; MAPK: Mitogen Activated Protein Kinase; Hsp90: Heat Shock Protein 90; MPNST: Malignant Peripheral Nerve Sheath Tumour; MYCN: Myc Proto-Oncogene Protein; B-ALL: B-Cell Acute Lymphoblastic Leukemia; p21: Cyclin-Dependent Kinase Inhibitor; PI3K: Phosphoinositide 3Kinase; SRC: Family of Proto-Oncogenic Tyrosine Kinases; STAT: Signal Transducer and Activator of Transcription; EGFR: Epidermal Growth Factor Receptor; HER2: Human Epidermal Growth Factor Receptor 2; ER: Estrogen Receptor; OBHS: Oxabicycloheptene Sulfonate; EMT: Epithelial-Mesenchymal Transition; ZEB1: Zinc Finger E-Box Binding Homeobox 1; BJAB: Burkitt's Lymphoma Cell Line; TYMS: Thymidylate Synthetase; Tip60: Histone Acetyl Transferase of the MYST Family; CBP: cAMP-Response Element-Binding Protein; EP300: Adenovirus Early Region 1A Binding Protein p300; MSL1: Male Specific Lethal 1; ALS: Amyotrophic Lateral Sclerosis; Agxt2l1: Alanine-Glyoxylate Aminotransferase 2-like 1; Sgk1: Serum / Glucocorticoid Regulated Kinase 1; Sult1a1: Sulfo Transferase Family 1A Phenol- Preferring Member 1; Tsc22d3: TSC22 Domain Family Member 3; MPP: 1-Methyl 4-Phenylpyridinium; XIAP: X-Linked Inhibitor of Apoptosis; GVHD: GraftVersusHostDisease; COPD: Chronic Obstructive Pulmonary Disease; FOXP: Forkhead Box P3 Protein; Treg cell: Regulatory T Cells; NFkB: Nuclear Factor 'Kappa-Light-Chain- Enhancer' of Activated B-Cells; MCP-1: Monocyte Chemotactic Protein 1; MIP- 1α+: Macrophage Inflammatory Proteins 1 Alpha; mRNA: Messenger Ribonucleic Acid; siRNA: Small Interfering Ribonucleic Acid; IL-1β: IL-6; IL-8 and IL-1β: Different Interleukins Functioning as Cytokines; CD8: Cluster of Differentiation 8; PCR: Polymerase Chain Reaction; Smad3: Small Ribonucleoprotein Particle Protein 3; TGF-β: Transforming Growth Factor β; LPS: Lipopolysaccharide; CLP: Cecal Ligation and Puncture; HIV: Human Immunodeficiency Virus; CD4: Cluster of Differentiation 4; EBV: Epstein Barr Virus; CVB3: Coxsackievirus B3
Introduction
Today medical research is focused on ‘personalized medicines’ for the treatment of cancer, neurological disorders, inflammatory diseases and viral infections. Nowadays more and more links are described between genomic and genetic variations and their associated human diseases thus leading to a greater understanding of their aetiology. This new knowledge can then lead to new therapeutic avenues with a strong molecular rationale. Epigenetic aberrations often contribute essentially to the onset and progression of the above mentioned human diseases via the loss or gain of function of epigenetic regulatory proteins [1]. In this article, we review the recent clinically relevant developments in the field of histone deacetylases (HDAC) inhibitors regarding human diseases that are linked to abnormal expression and/or function of HDACs. Over 1,750 proteins in human cells can be posttranslationally modified at lysine residues via acetylation and deacetylation [2].
These modifications play a crucial role in the modulation of the epigenetic indexing system through targeting transcriptionally active chromatin domains. Two important families of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs) emerged to influence this highly dynamic system [3]. HATs add acetyl groups to the lysine residues of histones (as well as other acetyl-lysine-containing proteins) to neutralize their positive charges subsequently leading to a relaxed chromatin structure, which is more accessible to the transcription machinery. In contrast HDACs catalyse the deacetylation from histones (and other acetyl-lysine-containing proteins), leading to chromatin condensation and transcriptional repression [4, 5]. Nowadays these deacetylating enzymes are considered as valuable targets to treat aberrant deacetylation related to cancer but also various other diseases such neurological disorders, inflammation and viral infections reverse [6-8].
HDACs are expressed in all eukaryotic cells, and their activity is essential for cell proliferation, differentiation and homeostasis. Eighteen different human HDACs have been described in the literature so far. Their classification is based on their homology to yeast HDACs. So far 5 classes have been described with differences in structure, enzymatic activity, localization and tissue expression patterns. Eleven out of these eighteen enzymes possess highly conserved deacetylase domains and are zincdependent. To these classes belong: class-I (HDAC1, 2, 3 and 8, yeast RPD3 homologues); class-IIa (HDACs 4, 5, 7 and 9), class- IIb (HDACs 6 and 10 both with two catalytic sites) and class-IV (HDAC11 possessing conserved residues shared with both class-I and class-II deacetylases).
In more detail class-I HDACs, being expressed in all tissues, show significant HDAC activity primarily in the nucleus where they are present in multi-protein complexes with transcription factors and co-repressors [4]. Differently, class-IIa HDACs are tissuespecific endowed with a low enzymatic activity, having low basal activities with acetyl-lysines. Furthermore, they might be efficient processors of specific, to date unknown, natural substrates [9]. Class-IIb HDACs have primarily non-epigenetic functions regulating protein folding and turnover [10]. In addition to the already mentioned so called “classical HDAC classes”, class-III HDACs are better known as sirtuins (SIRT1-7). These enzymes are all NAD+-dependent deacetylases possessing a structural relation to yeast protein Sirtuin 2. Their biological relevance and functions have recently been well reviewed [11,12].
HDAC inhibitors (HDACi) are mostly studied as anticancer agents, but there is a growing body of literature ascribing these enzymes to play a crucial role in other diseases such as neurological disorders, inflammatory processes and viral infections [13,14].
The pharmacophore of HDACi possesses usually the following well-known components: a cap group (CAP) allowing to interact with the rim of the catalytic tunnel of the enzyme, a polar connection unit (CU, apparently dispensable for HDAC8 selective inhibitors) connecting to a hydrophobic spacer (HS) enabling the inhibitor to lie into the aforementioned tunnel, and a Zn-binding group (ZBG) able to complex the Zn2+ at the bottom of the enzyme cavity [15].
To date numerous different CAP, CU, HS and ZBG combinations as HDACi have been either isolated from natural sources or synthesized displaying varying target specificity, pharmacokinetic properties and activity in laboratory [16] and clinical settings [17].HDACi can induce a range of cellular and molecular effects influencing various human diseases via hyperacetylation of histone and non- histone HDAC substrates. In the present review we would like to highlight their therapeutic potential, mainly in cancer; however, other potential therapeutic applications of these inhibitors in neurological diseases, infection and inflammation are also presented.
HDAC Modulation in Cancer Therapy
HDAC inhibition is mostly known and studied for its anti-tumoural effects such as cell death. There is large experimental proof in the present literature displaying a direct link between anti-tumour efficacy and apoptosis induction by HDACi [18]. The biological relevance and possible mechanisms of HDACi have recently been well reviewed [17,19].
Clinical development of HDACi
To date just a few HDACi have been approved by the FDA: vorinostat 1 (Zolinza®; Merck) for the treatment of refractory cutaneous T-cell lymphoma (CTCL) [20], romidepsin 2 (Istodax®; Celgene) for the treatment of CTCL and peripheral T-cell lymphoma (PTCL) [21], and belinostat 3 (Beleodaq®; Spectrum Pharmaceuticals) for the treatment of PTCL [17]. In early 2015 oral panobinostat 4 (Farydak®, Novartis) has been approved by the FDA, as combination therapy with bortezomib and dexamethasone in patients with recurrent multiple myeloma who have received at least two prior treatment regimens, including bortezomib and an immunomodulatory agent [22], being currently studied also for other types of cancer.
More than 350 clinical trials [23,24] have been carried out or are on-going involving HDACi not only as single therapeutic [25] but also in combination with other targeted agents [26] against various human diseases. These trials are often focused on haematological malignancies [27] and carried out in pretreated patients in a refractory state of their disease possibly explaining the poor efficacy seen in many trials [28] supporting the hypothesis, that an intact immune system potentially crucial for HDACi response in patients [29].
However, the phase 1 study of romidepsin 2 plus the tyrosinkinase inhibitor erlotinib in advanced non-small cell lung cancer [30] as well as the phase I study of the HDAC inhibitor vorinostat and the mTOR inhibitor ridaforolimus in advanced renal cell carcinoma possess both promising potential for further clinical development [31].
In the recent literature it is becoming evident that two opposite HDACi drug design strategies are applied: firstly, the development of highly selective inhibitors and secondly the development of dual- or multi-target inhibitors. The former method cannot only be used to elucidate the function of individual HDAC but also lead to novel potential inhibitors with fewer side effects [8]. The latter approach has promising potential for the antitumor therapy based on HDACi.
Effects of HDACi in combination with other chemotherapeutics
HDACi have been and are currently evaluated in combinations with other therapeutic agents with varying degrees of success [32]. An important successful approach lies in the combination of HDACi with DNAdamaging chemotherapeutics [33]. For example, the vorinostat 1 combined with idarubicin and cytarabine in nonpre- treated patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) led to a remarkable response rate of 85% [28]. Furthermore, HDACi have successfully been combined with DNA methyltransferase inhibitors (DNMTi) [33]. However, it needs to be elucidated whether the addition of HDACi such as valproic acid (VPA) 5 actually improves the efficacy of DNMTi [34]. To address this question, two large Phase II trials are currently ongoing; one is assessing the efficacy of 5azacytidine with and without entinostat 6 for the treatment of MDS, chronic myeloid leukaemia and AML [35] and the other is decitabine with or without VPA 5 and / or all-trans retinoic acid (ATRA), which in vitro display a very promising synergism, for AML treatment in Phase II after a successful Phase I [36].
The triple combination of the DNMTi decitabine, EZH2i 3-Deazaneplanocin-A (DZNep) and TSA 7 induced a remarkable synergistic antineoplastic effect against human AML cells via the reactivation of tumour suppressor genes (TSGs) in HL-60 and AML-3 leukemia cells. This triple combination was more effective than the combination of two agents or a single agent [37].
Additionally, VPA 5 is currently evaluated in Phase II with paclitaxel as second-line therapy for patients with advanced gastric cancer [38].
A further rational combination is involving an HDACi with a LSD1 inhibitor. The combination of LSD1inhibitor SP2509 and pan- HDACi panobinostat 4 was synergistically lethal against cultured and primary AML blasts exhibiting no toxicity. This lethal effect may be due to an increased induction of levels of the proapoptotic proteins p27 and BIM in AML cells exposed to cotreatment with SP2509 and 4, as compared with each single drug alone consequently providing a promising potential AML therapy option [39].
Another promising attempt lies in the combination of HDACi with hormone therapy to repress the transcription and function of estrogen receptors and androgen receptors via modified acetylation patterns [40]. For example, promising preclinical results [33] are currently verified in a Phase II clinical trial of vorinostat 1 and tamoxifen in patients with hormone therapy resistant breast cancer with an overall response rate of 19% [40].
Based on positive preclinical studies [33], the antiandrogen agent bicalutamide is currently being studied together with vorinostat 1 and panobinostat 2 for the treatment of prostate cancer [35].
There is a growing body of literature of promising preclinical evaluations of HDACi combined with agents targeting apoptosis proteins. Taken into account that HDACi are known to modulate the expression of key apoptosis proteins [17], there is a strong rationale to combine these with death receptors agonists or prosurvival protein antagonists. For example, vorinostat 1 or panobinostat 4 at sublethal doses sensitize malignant tumour cells, but not healthy cells, leading to TNFrelated apoptosisinducing ligand (TRAIL) induced apoptosis in syngeneic murine models of breast cancer and adenocarcinomas [41].
Trichostatin A (TSA) 7 possesses a potent and specific inhibition activity of mammalian HDACs in vitro as well as in vivo. For example, 7 was studied in combination with the DNMTi azacytidine for the treatment of human multi-drug resistant osteosarcoma HosDXR150 indicating that the acquired growth and survival advantage of these malignant cells is abolished by simultaneous epigenetic inactivation of p53-independent apoptotic signals and osteoblast differentiation pathways [42]. Unfortunately, TSA 7 did not meet the high expectations regarding its activity in clinical trials, possibly due to its low biodistribution and rapid metabolism [43]. Despite this, Ranganathan et al. were able to demonstrate that TSA could prevent nephrotoxicity under cisplatin treatment via upregulation of the activated microglia / macrophage WAP domain protein (AMWAP) [44]. The pan-HDACi AR-42 8 has been shown to down regulate CD44, a glycoprotein likely to be associated with the lenalidomide / dexamethason treatment resistance in myeloma both in vitro (myeloma cells from patients) and in vivo (primary myeloma mouse model) [45].
Multiple myeloma cells are producing a large number of misfolded proteins that need to be eliminated through the HDAC6 regulated aggresome and the proteasome [46,47]. Dual inhibition of these two pathways is resulting in the accumulation of defective proteins and subsequent tumour cell death caused by proteotoxicity, thus explaining the mechanistic rational approach behind the combination of HDAC6 selective inhibitors and proteasome inhibitors [48].
Amengual et al. investigated the therapeutic impact of the selective HDAC6 inhibitor ACY-1215 9 in combination with the proteasome inhibitor bortezomib in xenograft mice of lymphoma with promising results representing an innovative and rational approach for an improved therapy [48].
Other encouraging results have been observed in preclinical combination studies of vorinostat 1 with daunorubicin (a topoisomerase II inhibitor [49], camptothecin (a topoisomerase I inhibitor) [50], bexarotene (a retinoid X receptor agonist) [51], lovastatin (a 3hydroxy3methylglutaryl CoA reductase (HMGR) inhibitor) [52], pazopanib (multi kinase inhibitor) [53] carfilzomib (a proteasome inhibitor) [54], as well as panobinostat 2 with MK- 1775 (a Wee1 inhibitor) [55] or azacytidine (a DNMTi) [56].
Studies of class / isoform selective HDACi
Thailandepsin A (TDP-A) 10 is a recently discovered depsipeptide class I HDAC inhibitor with broad anti-proliferative activities at low nanomolar concentrations via an intrinsic apoptotic pathway through increase of pro-apoptotic protein Bax, decrease of antiapoptotic Bcl-2, and cleavage of caspase-3 and poly (ADP-ribose) polymerase (PARP). In the MDA-MB-231 breast cancer xenograft model, TDP-A 10, packed in disulfide cross-linked micelles as a nanoformulation to improve targeting and safety, were more efficacious than the FDA-approved romidepsin at well-tolerated doses [57] (Figure 1).
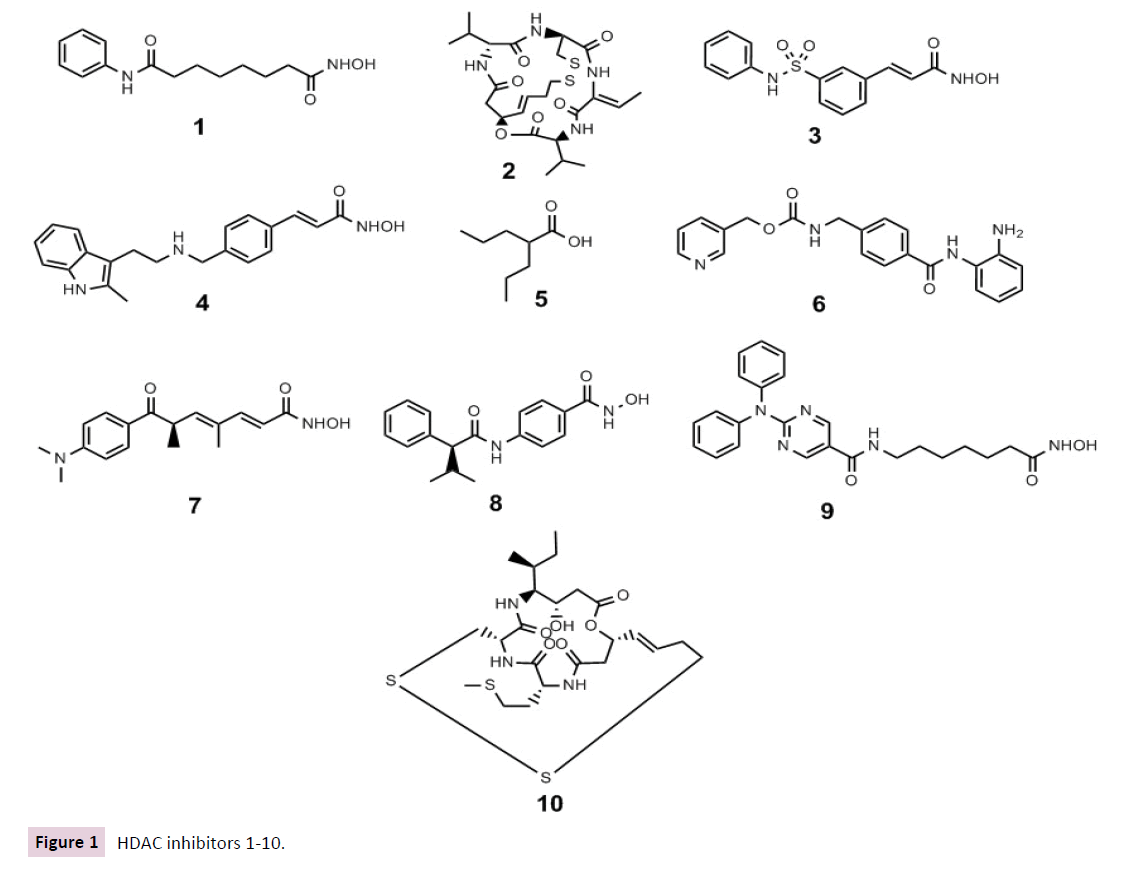
Figure 1 HDAC inhibitors 1-10.
Yao et al. described recently a successful structure-based design of novel cyclic depsipeptides that selectively target class I HDAC isoforms. They discovered compound 11 exhibiting more than 20-fold selectivity toward human cancer cells over human normal cells in comparison with romidepsin 2, demonstrating low probability for toxic side effects while demonstrating excellent in vivo anticancer activities in a human prostate carcinoma (Du145) xenograft model [58].
Compound 12 is a potent and selective class I HDAC inhibitor, which has been shown to possess anti-proliferative and cytotoxic activity in colorectal cancer cells arresting them at the G2 / M phase. The anti-tumour activity and underlying molecular mechanisms of compound 12 were further confirmed using the HCT116 xenograft model in vivo suggesting the further development of this compound [59].
Specific class IIa HDAC inhibitors have been described just recently. YK4272 13 is able to inhibit the nuclear import of II HDACs thus leading to their inhibition [60]. Notably, 13 were observed to restrict tumour cell growth in vivo [60], supporting the hypothesis that nuclear class II HDACs could be essential for, or at least augment, the enzymatic activity of nuclear class I HDACs. Class IIa HDACs are well described to possess a much weaker deacetylasing activity than class I HDACs when assessed with standard acetyllysine assay [9]. Class IIa HDACs, in contrast to class I or class IIb HDACs, can interact with trifluoroacetyllysine substrates indicating that the natural substrate or substrates for class IIa HDACs need still to be unrevealed rather than hypothesize a low catalytic activity [9].
The HDAC3selective inhibitor RGFP966 14 is inhibiting CTCL cell growth in vitro as well as DNA replication. The administration of this drug led to a significant reduction in DNA replication fork velocity within the first hour of drug treatment via the inhibition of HDAC3; thus disrupting DNA replication of the rapidly cycling tumor cells, in the end leading to tumour cell death. Importantly, these molecular and biological findings were confirmed via siRNAmediated knockdown of HDAC3, confirming on a genetic level the HDAC3selective effects of this compound [61]. The effectiveness of 14 has been recently confirmed also in AML, Eμ-Myc lymphoma and acute promyelocytic lymphoma (APL) [62]. T247 15 displayed potent selective HDAC3 inhibition with a submicromolar IC50 inducing a dose-dependent selective increase of NF-kB acetylation, a known target of HDAC3, in human colon cancer HCT116 cells leading to growth inhibition [63].
HDAC6 is able to inhibit cancerous Hsp90 chaperone activities by disrupting Hsp90 / p23 interactions. Compound 1A12 16 was identified to disrupt the aforementioned interactions in tumour xenografts mice via HDAC6 inhibition in a dose dependent manner [64]. Seidel et al. identified compound 17, a 4-hydroxybenzoic acid, as selective HDAC6 inhibitor reducing proliferation, colony and spheroid formation as well as viability of prostate cancer cells via α-tubulin acetylation resulting in remodeling of microtubular organization and via increasing HSP90α acetylation levels resulting in a decreased HSP90α regulation of the human androgen receptor in prostate cancer cells [65]. Recently quinazoline has been described as a new CAP group driving the selectivity towards HDAC6 inhibition. Compound 23bb 18 displayed a superior tumour reduction in vivo efficacy evaluations of colorectal HCT116, acute myelocytic leukemia MV4-11, and B cell lymphoma xenograft mice, then ACY-1215 at the same dose [66].So far not too many HDAC8specific inhibitors such as PCI34051 19 and C149 20 have been described. Both aforementioned compounds led to a decreased proliferation and induction of apoptosis in T cell leukaemia and lymphoma very likely targeting cohesin, a protein complex that regulates the separation of sister chromatids during cell division [67]. A malignant peripheral nerve sheath tumour (MPNST) is an aggressive sarcoma that is notoriously therapy-resistant. Lopez et al. demonstrated that the HDAC8i 19 induced cell growth inhibition and marked S-phase cell cycle arrest in human and murine-derived MPNST cells thus potentially leading to a MPNST treatment [68]. Also in neuroblastoma 19 exhibited as selective HDAC8i antineuroblastoma activity via downregulation of MYCN oncogene expression without toxicity in two xenograft mouse models of MYCN oncogene-amplified neuroblastoma [69].
In contrast, increased cytoplasmic HDAC8 immunoreactivity was associated with an improved survival from diagnosis of primary melanoma thus HDAC8 might be a prognostic biomarker in melanoma [70]. However, it should be noticed that the detailed physiological properties and functions of HDAC8 are still not yet fully understood [67]; despite that, the rationale use of HDAC8selective inhibitors in cancer but also in human diseases such as parasitic and viral infections, as well as neurodegenerative diseases is emerging [71] (Figure 2).
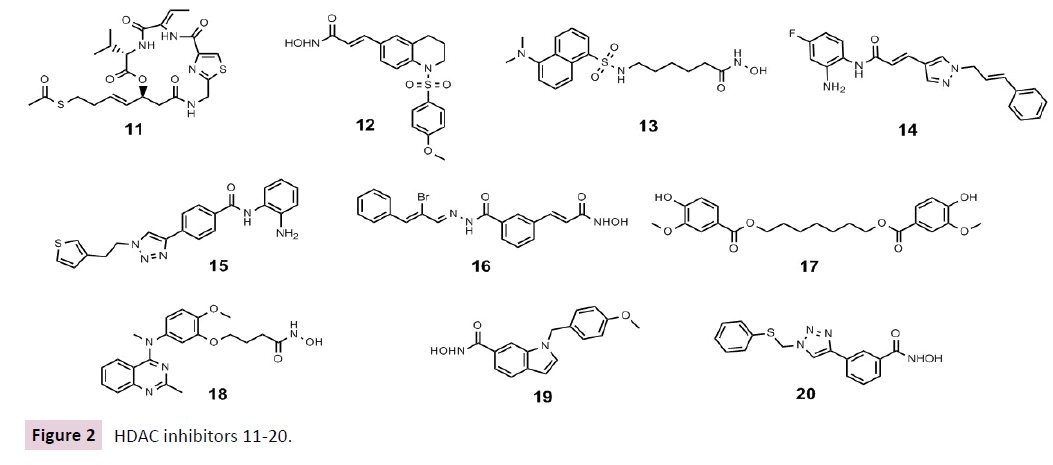
Figure 2 HDAC inhibitors 11-20.
Due to their 95% similarity within the catalytic binding domain, isoform selective inhibitors of HDAC1 or HDAC2 are still a challenge for medicinal chemists [72] Nevertheless compounds that preferentially inhibit both of these isoforms have been discovered recently. MRLB223 21, as an HDAC1 and HDAC2 selective inhibitor, led to link histone hyperacetylation in vivo displaying antitumour effects using the same apoptotic pathways as vorinostat 1 via the intrinsic apoptotic pathway independently of p53 [73]. Recently Stubbs et al. demonstrated via shRNAmediated knockdown of HDAC1 or HDAC2 and the chemical probe compound 60 22 that a specific inhibitor of HDAC1 and HDAC2 could be therapeutically useful for patients with B-cell acute lymphoblastic leukemia (B-ALL) [74].
Recently our research group discovered an 1,3,4-oxadiazolecontaining 2-aminoanilide (3i) 23 as a selective HDAC1 inhibitor over isoform 4 and 6. With western blot experiments we were able to display that (3i) 23 increased both histone H3 and α-tubulin acetylation and led to p21 induction, being more efficient than vorinostat 1. In this study could be proven in several AML cell lines, as well as in U937 cells also in combination with doxorubicin, that compound 23 is slightly better than vorinostat 1 [75].
HDACi as multi-targeting drugs
Polypharmacological HDACi do not inhibit only HDACs but involve at least one other target. Kasparokova et al. developed an innovative prodrug containing a photoactivatable platinum (IV) complex conjugated with suberoyl-bis-hydroxamic acid 24 in highly active in A2780 tumour cells. The conjugate exerts, after selective photo activation in and around a tumour, thereby increasing selectivity towards cancer cells, two functions: on the one hand activity as a platinum (II) anticancer drug and on the other hand histone deacetylase (HDAC) inhibitor activity. These results suggest that this strategy is a valuable novel approach to photodynamic rational anticancer therapy [76].
For example, CUDC907 25 containing a hydroxamic acid and a morpholinopyrimidine to inhibit both HDACs and phosphoinositide 3kinase (PI3K) [77], is currently evaluated in Phase I for the treatment of lymphoma and multiple myeloma (NCT01742988). This compound can downregulate and suppress the activation of the SRC / STAT signalling pathway and multiple receptor tyrosine kinases, again presumably because of its HDAC inhibitory activity as we observed that panobinostat 4 also induced a similar effect exhibiting greater growth inhibition and proapoptotic activity than single-target PI3K or HDAC inhibitors in both cultured and implanted cancer cells [77].
Another interesting compound in clinical trials for solid tumours is CUDC101 26. This compound inhibits beside HDACs, the tyrosine kinases EGFR and HER2 combining a hydroxamic acid with a phenylaminoquinazoline (NCT01702285, NCT01171924, NCT01384799 and NCT00728793) [23, 78, 79].
In acute promyelocytic leukemia (APL), the promyelocytic leukemia gene (PML) is influenced by an HDAC-containing complex with the retinoic acid receptor α (RARα). MC2392 27 is binding to the aforementioned moiety and is selectively inhibiting the HDACs resident in this repressive complex responsible for the transcriptional impairment in APL [80].
Recently Tang et al. developed a series of dual-acting ER and histone deacetylase inhibitors. These novel hybrid compounds combine an indirect antagonism structure motif of estrogen receptor (ER) (OBHS, oxabicycloheptene sulfonate) with the HDAC inhibitor vorinostat 1. These non -toxic OBHS-HDACi conjugate 28 exhibited good ER binding affinity and excellent ER-α antagonistic as well as good HDAC1 and 6 inhibitory activities, being more potent than tamoxifen and also possessing selective anticancer activity being more potent against MCF-7 breast cancer cells than DU 145 prostate cancer cells [81].
Aberrant activation of an epithelial-mesenchymal transition (EMT) and an associated cancer stem cell phenotype such as pancreatic cancer are considered to be a major cause of therapy resistance. Particularly, the EMT-activator ZEB1 was shown to confer stemness and resistance. The known class I HDAC inhibitor mocetinostat 29 is capable to interfere with ZEB1 function, restore the expression of a major chemotherapy sensitizer miR- 203, repress stemness properties, and induce sensitivity against chemotherapy being another possible approach for combinations of selected epigenetic drugs with standard chemotherapy for the rational treatment of aggressive solid tumours, such as pancreatic cancer [82].
Huang et al. demonstrated that MPT0E028 30 exhibits a potent dual function of HDAC and Akt inhibition, leading to B-cell lymphoma apoptosis in vitro and in vivo. Its antitumor activities include inducing apoptosis, HDAC inhibition, Akt pathway deactivation, and in vitro regulation of many genes, such as TP53, MYC, STAT family, and prolongation of survival rate and reduction of tumour volume in vivo in a BJAB xenograft model [83] (Figure 3).
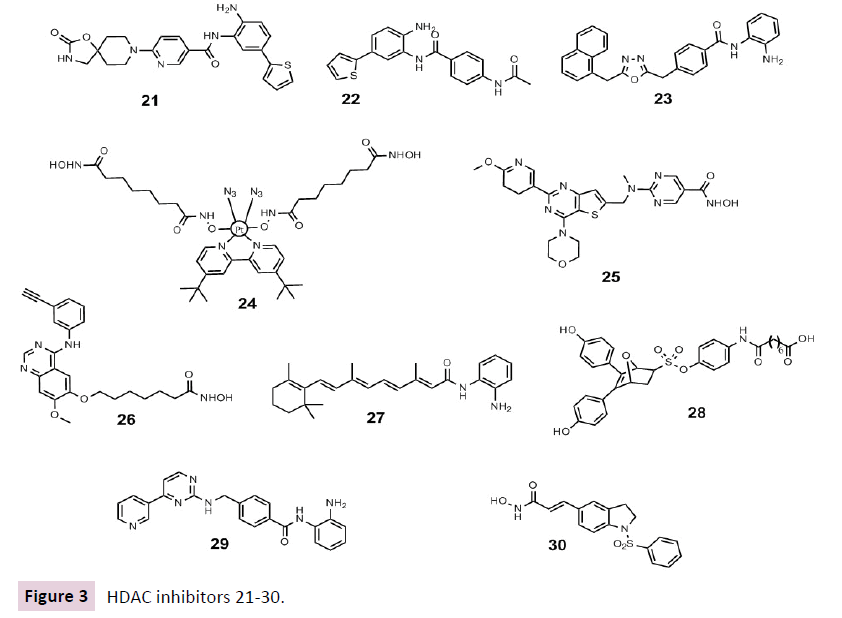
Figure 3 HDAC inhibitors 21-30.
Genotoxic drugs constitute a major treatment modality for human cancers; however, cancer cells' intrinsic DNA repair capability often increases the threshold of lethality and renders these drugs ineffective. CY190602 31 is a novel bendamustine-derived drug with anticancer potency in vitro and in vivo which can be attributed to its ability to and inhibit HDACs and to influence DNA repair. The authors used this novel DNA / HDAC dual-targeting drug as a tool, to explore the HDAC's role in DNA repair. They found that HDAC activities are essential for the expression of several genes involved in DNA synthesis and repair, such as TYMS, Tip60, CBP, EP300, and MSL1. These findings provide rationales for incorporating HDAC inhibitory moieties into genotoxic drugs, so as to overcome the repair capacity of cancer cells possibly improving cancer therapy in the years to come [84].
Applications of HDACi in Inflammation
Enhanced Treg cell number and function after HDACi administration in rodent models of inflammatory diseases such as arthritis, inflammatory bowel disease, hypertension, septic shock, colitis and graftversushostdisease (GVHD) encouraged deeper studies on HDAC role in inflammation. During these studies class II, class IIaor isoformspecific inhibitors merged as the best candidates for the treatment of inflammatory diseases [85, 86]. In contrast pan-HDACi are possibly compromising host defence aggravating atherosclerosis as well as COPD [87, 88], through inhibition of class I HDACs, which are key repressors of proinflammatory cytokines. PanHDACi such as TSA 7 and vorinostat 1, but not class Ispecific HDAC inhibitors like MS275 6 did not lead to the development of colitis and promoted its resolution in rodent models via FOXP3 (+) Treg cells modulated through class II specific HDACi [89]. Preclinical studies revealed the effectiveness of HDAC inhibitor MPT0G009 32 in inflammatory arthritis, suggesting it for clinical development in this setting. 32 inhibited cytokine secretion and macrophage colony-stimulating factor/ receptor activator of NFkB ligand-induced osteoclastogenesis by macrophages. Overexpression of HDAC 1 and 6 in cells diminished these effects, suggesting that these outcomes were induced by the inhibition of their activity [90]. The implication of HDAC in rheumatoid arthritis (RA) was further confirmed by two independent studies published in 2015. First Cantley et al. investigated the novel HDAC1 selective inhibitor, NW-21 33. 33 and entinostat 8 suppressed osteoclast formation and activity and reduced MCP-1 and MIP-1+ mRNA expression in monocytes via HDAC1 inhibition leading to reduced inflammation and bone loss in the arthritis model [91]. Later Hawtree et al. demonstrated how siRNA-mediated HDAC1 knockdown in a collagen-induced arthritis model resulted in reduced joint swelling, cartilage and bone damage and lower TNF levels in joint tissue. These results may implicate that HDAC1 is an important mediator of tissue damage in RA with a potential therapeutic application of its inhibitors. In particular, selective HDAC1 inhibitors, such as NW- 21 33 and entinostat 8, may be useful for treating RA; as such drugs can simultaneously target both inflammation and bone resorption [92].
The specific class I HDACs inhibitor sodium butyrate 34 exerts potent anti-inflammatory effects in the treatment of acute gout decreasing the production of IL-1β, IL-6 and IL-8 [93].
Inflammation and fibrosis are implicated in the pathogenesis of hypertensive kidney damage. Choi et al. reported that the selective inhibition of HDAC6 with tubastatin A 35 in a hypertensive rat model, prevents fibrosis and inflammation as determined by quantitative real-time PCR, western blot, and immunohistochemistry. Interestingly, the combination of the HDAC6 inhibitor and Smad3 knockdown synergistically blocked transforming growth factor β (TGF-β) or ANG-induced fibrosis. These results suggest that HDAC6 may be a valuable therapeutic target also for the treatment of hypertension-induced kidney fibrosis and inflammation [94].
Between 2010 and 2013 the Li et al. reported vorinostat 1 as able to improve survival in rodent models of lipopolysaccharide (LPS) induced endotoxemia and cecal ligation and puncture (CLP) induced septic shock. Vorinostat 1, as panHDACi, also increased host vulnerability to bacterial infection. Hence they hypothesised that the use of isoform selective inhibitors could lead to a focused activity. More recently they demonstrated that in a lethal mouse model of cecal ligation and puncture (CLP) HDAC6 inhibition by Tubastatin A 35 significantly improves animal survival, reduces "cytokine storm" attenuates acute livery injury, increases bacteria clearance and immune cell phagocytosis, and inhibits macrophage apoptosis. In this regards, isoform selective inhibitors can be considered a route straightforward to reduce side effect and improve efficiency [95].
ACY-1215 36, an HDAC6 inhibitor, prevented the development of contact hypersensitivity and GVHD-like disease in vivo by modulating CD8 T-cell activation and functions [96]. Additionally, HDAC6, as a key modifier of T-cell receptor signalling, is highly expressed on CD8 T cells and has been shown to regulate immune responses through interactions between T cells and antigenpresenting cells. ACY-1215 36 prevented the development of contact hypersensitivity CHS and graft-versus-host disease GVHD-like disease in vivo by modulating CD8 T-cell activation and functions in a murine CD8 T cell-related skin disease model. Interestingly ACY-1215 in vitro revoked the differentiation of naïve CD8 T-cells to effector T-cells through anti-CD3 / CD28 antibody– or antigen–specific stimulation and blocked the mitogen activated protein kinase (MAPK) pathway after increasing the binding of the acetylated HSP90 to lymphocyte specific protein tyrosine kinase and disrupting the lymphocyte-specific protein tyrosine kinase phosphorylation [96].
In 2014 ITF3056 37 an HDAC8-selective inhibitor analogue of Givinostat, was described for its anti-inflammatory activity. ITF3056 downregulated pro-inflammatory cytokines gene expression and consequently their production in vitro in human blood monocytes, as well as in vivo in C57BL/6 mice [97]. Recently, based on these data MC2625 (HDAC3/6/8 inhibitor) 38 and MC2780 (HDAC6 inhibitor) 39 down regulated IL-1β and IL-6 mRNA expression and promoted IL-10 transcription with no effects on cell viability at anti-inflammatory dosages once tested in LPS-induced inflammation in KB31, C2C12 and 3T3-J2 cell lines. Tested in C57BL/6J female mice those compounds 38 and 39 were able to reduce host inflammation in silicone implants, thank to IL- 10 increased expression and IL-1β / IL-6 reduced production [98].
Current clinical trials of HDAC inhibitors addressing immune modulation and inflammation are focused on sickle cell disease using pan-HDACi vorinostat 1 or panobinostat 4 (NCT01245179 and NCT01000155) [23], in which inflammation mainly contributes to the disease pathology. The treatment and / or prevention of GVHD in the context of stem cell transplantation using the same above mentioned drugs 1 and 4 is another area of current important in clinical evaluation (NCT00810602, and NCT01111526) [23].
HDACi as a Therapeutic Strategy for Neurological Disorders
HDACi are used in the treatment of neurological diseases since many years, even before the molecular targets of these drugs have been described. VPA 5 was approved by the FDA in 1978 as an anticonvulsant drug for the treatment of absences as well as partial and generalized seizure disorders [99]. HDACi possess promising activities in various animal models of neurodegenerative diseases, such as Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, ischaemic stroke, amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (reviewed in [100, 101]).
Suo et al. Studied the effects of HDAC inhibition on neuronrestrictive silencer factor NRSF-mediated repression in Parkinson using the pan HDACi TSA 7. A single dose TSA pre-treatment upregulated the expression of tyrosine hydroxylase and brain-derived neurotrophic factor (BDNF) and protected the nigrostriatal dopaminergic pathway against MPTP (1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine)-induced degeneration in wild type mice. However, the protective functions of 7 were fully abolished in NRSF neuronal deficient mice suggesting that it serves as an essential mediator for the neuroprotection of Parkinson [102].
However, pan hydroxamatebased HDACi can lead to neurotoxic effects under oxidative stress conditions via glutathione depletion [103], whereas in contrast the HDAC6specific inhibitor tubastatin A 35 displayed neuroprotective properties [104]. Furthermore, the specific inhibition of HDAC6 tubastatin A 35 or ACY-1215 36 led to decreased levels of pathogenic tau protein ameliorating clinical particularities of tauopathies such as Alzheimer’s [105]. Both aforementioned compounds 35 and 36 increased tubulin acetylation, mediated by HDAC6, as well as reduced the production and facilitated the autophagic clearance of Abeta and hyperphosphorylated tau. However, despite their auspicious promising initial data, these findings need to be studied deeper, as HDAC6 might display both neurodegenerative and neuroprotective effects [106].
As explained above, the development of isoform selective inhibitors of HDAC1 or HDAC2 are a challenge; despite that, compound 60 22 were recently described to quite selectively inhibit both of these class I HDACs [107]. 22 as HDAC1/2 selective inhibitor led mood stabilization and antidepressant effects in a mouse model via elevated levels of acylated H4K12ac thus leading to an increase of the following various mood related proteins: alanine-glyoxylate aminotransferase 2-like 1 (Agxt2l1), serum/ glucocorticoid regulated kinase 1 (Sgk1), sulfo- transferase family 1A phenol-preferring member 1 (Sult1a1), and TSC22 domain family member 3 (Tsc22d3) [107]. Interestingly vorinostat could not influence positively the moodrelated behavioural effects as 22 did, as well as lead to the aforementioned changes in gene expression observed in the brains of mice. Regarding the other previously mentioned HDAC1/2 selective compound MRLB223 21 with comparable antitumour effects to vorinostat 1, the true specificity and selectivity of HDAC inhibitors and the differential effects of small molecules on HDACs in the recombinant, purified form compared to physiological conditions, needs to be questioned thus carefully studied in the years to come [73, 107](Figure 4).Wagner et al. described very recently the HDAC2 selective inhibitors BRD6688 40 and BRD4884 41. These two compounds display not only promising activities in Alzheimer’s treatment but could serve also as lead structures for the development of isoform selective HDACi. Furthermore, 40 and 41 might be used to probe the biological functions of the different HDAC isoforms studying their relevance in various diseases [72]. Both compounds increased H4K12 and H3K9 histone acetylation in primary mouse neuronal cell culture assays, in the hippocampus of CK-p25 mice, a model of neurodegenerative disease, and rescued the associated memory deficits of these mice in a cognition behavioural model.
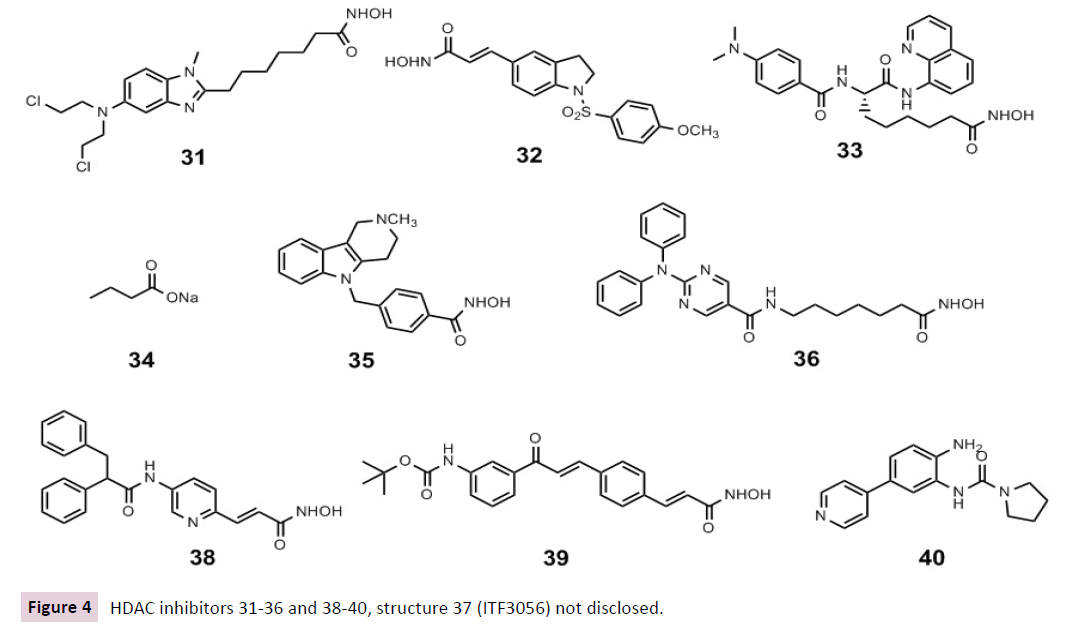
Figure 4 HDAC inhibitors 31-36 and 38-40, structure 37 (ITF3056) not disclosed.
Orally administered vorinostat 1 can ameliorate memory impairment in mouse models of age-associated cognitive decline and amyloid deposition without leading to massive and unspecific changes in transcriptome plasticity. As stated above the mechanistic insights on the action of HDACi in the brain are poorly understood, consequently HDACi are not yet studied for the treatment of Alzheimer’s in clinical trials [108].
There is convincing evidence suggesting that HDAC4 plays a central role in the brain physiology and that it is deregulated in several neurodegenerative disorders such as Alzheimer’s, Huntington, Parkinson, therefore representing a suitable therapeutic target, through which HDACs inhibition may occur [109]. Crucially, relatively little is known regarding individual HDACs functions in the adult brain. Recent studies identified HDAC4 as a critical component of several neurological processes including neuronal survival and synaptic plasticity in healthy and diseased brains. However, little is known about HDAC4 cellular process like: mechanisms governing HDAC4 cellular localization, posttranslational modification and a proteolytic cleavage, especially in the diseased brains. In addition, HDAC4 transcriptional regulation has not been studied and therefore the description of the specific transcription factors and regulatory elements driving HDAC4 expression should be carefully undertaken [109]. A first attempt in this direction was the proof of concept study by structure based from Bürli et al. design resulting in compound 42 being active in Huntington disease. However this compound is not fully HDAC4 selective but class IIa selective over I and IIb making further optimization and studies necessary [110].
Choong et al. identified a novel HDAC1/2 isoform-specific inhibitor, K560 43, with protective effects against neuronal death in both in vitro and in vivo Parkinson's disease model. K560 37 attenuated cell death induced by MPP(+) in differentiated SHSY5Y cells through the sustained expression of an anti-apoptotic protein, X-linked inhibitor of apoptosis (XIAP). Inactivation of MAPK cascades, reduced p53 phosphorylation, and downregulation of p53-upregulated modulator of apoptosis on K560 43 treatment were also observed. Furthermore, pre- and postoral administration of K560 to C57BL/6 mice prevented the loss of dopaminergic neurons in substantia nigra, suggesting that selective inhibition of HDAC1 and HDAC2 by 43 may pave the way to new strategies for Parkinson's disease treatment [111].
However, the development of panHDACi to treat neurological diseases should proceed with caution as these inhibitors could lead to off-target as well as severe side effects in neurobiology and neurological diseases [112].
HDACi as Tools to Fight Viral Infections
HDACi have been also evaluated for their potential in antiviral therapeutic strategies. We recently summarized HDAC inhibitors as able to fight human immunodeficiency virus (HIV), being able to eradicate latent virus reservoirs. Notwithstanding the best known [113] and even clinically studied hydroxamate-based HDACi vorinostat 1, romidepsin 2 and panobinostat 4 and are currently being tested in combination with various antiretroviral therapies (NCT01680094, NCT01319383, NCT02336074, NCT02092116, NCT02471430, NCT02475915) [23], one non-hydroxamate has been newly discovered. In 2015, Badia et al. described the new thiol based nanomolar pan-HDAC inhibitor ST7612AA1 44 as a potent in vitro inducer of latent HIV reactivation at subnanomolar concentrations. Its efficiency was comparable to that of panobinostat or superior to vorinostat 1. ST7612AA1 44 -induced reactivation was not affected by co-treatment with known antiretrovirals and their activity was not affected by 44, suggesting a possible co-administration to have a dual and concerted activity for a “shock and kill” strategy. 44 efficiency was not due to activation or proliferation of CD4+ T cells and it caused lower cell toxicity than romidepsin [114].
The previously mentioned selective HDAC3 inhibitor T247 15 was demonstrated to activate HDAC3 controlled HIV-1 gene expression in latent HIV-infected cells [63].
The selective inhibition of HDAC4 using MC1568 45 might represent a unique strategy to modulate the expression of therapeutic viral vectors, as well as of integrated HIV-1 proviruses in latent reservoirs without displaying significant cytotoxicity [115].
In recent times Husain et al. demonstrated that HDAC6 inhibition via tubacin 46, a domain-specific inhibitor able to bind to one of the two HDAC6 catalytic domains affected influenza virus functions through the negative regulation of the trafficking of viral components to the site of influenza virus via the HDAC6 substrate, acetylated microtubules [116] (Figure 5).
Figure 5 HDAC inhibitors 41-46.
Even though HDACi might be important for future treatments eradicating viruses such as HIV, the risk of activating a latent virus in infected patients when given an HDACi for another indication should be still studied further in detail. For example, latent hepatitis B virus and EBV infections were reactivated in patients suffering from cancer under treatment with romidepsin 2 [117].
Interestingly in 2015 Zhou et al., based on the antiviral efficiency of HDACi, evaluated the therapeutic application of vorinostat 1 or TSA 7 in the treatment of viral myocarditis that is mainly caused by coxsackievirus B3 (CVB3). Surprisingly they found that treatment of CVB3-infected BALB/c mice with HDACi worsened the CVB3-caused myocarditis with an increased CVB3 replication and subsequently myocardial apoptosis. Conversely, ribavirin inhibition of CVB3 replication significantly reversed the HDACiaggravated viral myocarditis. On the basis of these results any new application of HDACi as antiviral needs to be evaluated carefully. Moreover, possible adverse effects need to be taken in account in case of HDACi treatment in patients coinfected with CVB3s [118] .
Conclusion
In this review we highlighted that the growing understanding of specific HDACs in various disease aetiologies through the discovery of the molecular and biological alterations via HDAC inhibition in cancer but also non-cancer diseases such as neurological and inflammatory disorders as well as viral infections.
Most HDACi present in literature target multiple HDACs, which makes it rather difficult to assess whether their biological effects including clinical toxicities are due to the inhibition of a specific HDAC isoform, the combined inhibition of multiple HDACs and / or impacts on one or more multi protein complexes in which HDACs act as key enzymatic components [119]. Especially the latter point is underappreciated in present studies requiring further attention.
The development of selective HDACi is challenging due to the fact that the three-dimensional structures for most human HDAC isoforms are largely unknown. Despite this fact, some promising molecules presented in this review are the best known among the class and isoform selective HDACi.
In this review we presented some good examples of combination of HDACi with other epigenetic modulators and/or other chemotherapeutics. The way to a larger application of such combinations and a better understanding of their potential use is still long, and the reported examples, even if some are already in clinical trials, can be only seen as a starting point for deeper studies.
Although there is already a large panel of preclinical and clinical studies regarding HDACi, we believe that there is an urgent need of novel HDACi and novel innovative therapeutic approaches.
In our view a better understanding of the molecular and biological roles of distinct HDACs and HDACcontaining complexes and the underlying mechanisms of action of HDAC inhibitors including their off-target effects will certainly lead to a more specifically addressed development and rational use of HDAC inhibitors in the years to come.
Acknowledgement
This work was supported by the RF-2010-2318330 grant from the Italian Ministry of the Health, the Sapienza Ateneo Project 2015, the IIT-Sapienza Project, the FP7 Projects BLUEPRINT/282510 and A-PARADDISE/602080, and the COST-Action: EPICHEMBIO (CM1406).
Declaration of Interest
The authors declare no conflict of interest.
References
- Berdasco M, Esteller M (2013) Genetic syndromes caused by mutations in epigenetic genes. Hum Genet 132: 359-383.
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, et al. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325: 834-840.
- Clayton AL, Hazzalin CA, Mahadevan LC (2006) Enhanced histone acetylation and transcription: a dynamic perspective. Molecular cell23:289-296.
- Yang XJ, Seto E (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nature reviews Molecular cell biology 9:206-218.
- Haberland M, Montgomery RL, Olson EN (2009)The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature reviews Genetics10:32-42.
- Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature reviews Cancer6:38-51.
- Glozak MA, Seto E (2007) Histone deacetylases and cancer. Oncogene 26: 5420-5432.
- Zhang L, Han Y, Jiang Q, Wang C, Chen X, et al. (2015) Trend of histone deacetylase inhibitors in cancer therapy: isoform selectivity or multitargeted strategy. Medicinal research reviews 35: 63-84.
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, et al. (2007)Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proceedings of the National Academy of Sciences of the United States of America 104:17335-17340.
- Boyault C, Sadoul K, Pabion M, Khochbin S (2007) HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 26:5468-5476.
- Outeiro TF, Marques O, Kazantsev A (2008) Therapeutic role of sirtuins in neurodegenerative disease.Biochimicaetbiophysicaacta1782:363-369.
- Yao Y, Yang Y, Zhu WG (2014)Sirtuins: nodes connecting aging, metabolism and tumorigenesis. Current pharmaceutical design 20:1614-1624.
- Dinarello CA (2010) Anti-inflammatory Agents: Present and Future. Cell 140: 935-950.
- Gray SG (2011) Epigenetic treatment of neurological disease. Epigenomics3:431-450.
- Mai A, Massa S, Rotili D, Cerbara I, Valente S et al. (2005) Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Medicinal research reviews 25: 261-309.
- Johnstone RW (2002) Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 1:287-299.
- West AC, Johnstone RW (2014)New and emerging HDAC inhibitors for cancer treatment. JClin Invest 124: 30-39
- Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5: 769-784.
- Benedetti R, Conte M, Altucci L (2015) Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid Redox Signal 23:99-126.
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, et al. (2007) Phase 2 trial of oral vorinostat (suberoylanilidehydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood109:31-39.
- VanderMolen KM, McCulloch W, Pearce CJ, Oberlies NH (2011)Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J Antibiot (Tokyo) 64:525-531.
- Garnock-Jones KP (2015)Panobinostat: first global approval. Drugs. 75: 695-704.
- https://www.clinicaltrials.gov/
- Nervi C, De Marinis E, Codacci-Pisanelli G (2015) Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenetics 7:127.
- Nebbioso A, Carafa V, Benedetti R, Altucci L (2012) Trials with 'epigenetic' drugs: an update. MolOncol 6:657-682.
- Qiu T, Zhou L, Zhu W, Wang T, Wang J, et al. (2013) Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol 9:255-269.
- New M, Olzscha H, La Thangue NB (2012) HDAC inhibitor-based therapies: can we interpret the code? MolOncol 6:637-656.
- Garcia-Manero G, Tambaro FP, Bekele NB, Yang H, Ravandi F, et al. (2012) Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenousleukemia or myelodysplastic syndrome. J ClinOncol 30:2204-2210.
- West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, et al. (2013) An intact immune system is required for the anticancer activities of histone deacetylaseinhibitors. Cancer Res 73:7265-7276.
- Gerber DE, Boothman DA, Fattah FJ, Dong Y, Zhu H, et al. (2015) Phase 1 study of romidepsin plus erlotinib in advanced non-small cell lung cancer. Lung Cancer.
- Zibelman M, Wong YN, Devarajan K, Malizzia L, Corrigan A, et al. (2015) Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Invest New Drugs 33:1040-1047.
- Bots M, Johnstone RW (2009)Rational combinations using HDAC inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research15:3970-3977.
- Thurn KT, Thomas S, Moore A, Munster PN (2011) Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol7:263-283.
- Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, et al. (2007) Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J ClinOncol25:3884-3891.
- Valente S, Mai A (2014) Small-molecule inhibitors of histone deacetylase for the treatment of cancer and non-cancer diseases: a patent review (2011 - 2013). Expert OpinTher Pat 24: 401-415.
- Grishina O, Schmoor C, Dohner K, Hackanson B, Lubrich B, et al. (2015) DECIDER: prospective randomized multicenter phase II trial of low-dose decitabine (DAC) administered alone or in combination with the histone deacetylase inhibitor valproic acid (VPA) and all-trans retinoic acid (ATRA) in patients >60 years with acute myeloid leukemia who are ineligible for induction chemotherapy. BMC Cancer15:430.
- Momparler RL, Cote S, Momparler LF, Idaghdour Y (2014) Epigenetic therapy of acute myeloid leukemia using 5-aza-2'-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin Epigenetics 6:19.
- Fushida S, Kaji M, Oyama K, Hirono Y, Nezuka H, et al. (2015) Randomized Phase II trial of paclitaxel plus valproic acid vs.paclitaxel alone as second-line therapy for patients with advanced gastric cancer. Onco Targets Ther8:939-941.
- Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, et al. (2014) Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 28:2155-2164.
- Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, et al. (2011) A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. British journal of cancer 104:1828-1835.
- Martin BP, Frew AJ, Bots M, Fox S, Long F, et al. (2011) Antitumor activities and on-target toxicities mediated by a TRAIL receptor agonist following cotreatment with panobinostat. Int J Cancer 12811: 2735-2747.
- Capobianco E, Mora A, La Sala D, Roberti A, Zaki N, et al. (2014) Separate and combined effects of DNMT and HDAC inhibitors in treating human multi-drug resistant osteosarcoma HosDXR150 cell line. PLoS One9:e95596.
- Tan S, Liu ZP (2015) Natural Products as Zinc-Dependent Histone Deacetylase Inhibitors. ChemMedChem.
- Ranganathan P, Hamad R, Mohamed R, Jayakumar C, Muthusamy T, et al. (2015) Histone deacetylase-mediated silencing of AMWAP expression contributes to cisplatin nephrotoxicity. Kidney Int.
- Canella A, Cordero Nieves H, Sborov DW, Cascione L, Radomska HS, et al. (2015) HDAC inhibitor AR-42 decreases CD44 expression and sensitizes myeloma cells to lenalidomide. Oncotarget6:31134-31150.
- McConkey DJ, White M, Yan W (2012) HDAC inhibitor modulation of proteotoxicity as a therapeutic approach in cancer. Adv Cancer Res116:131-163.
- Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, et al. (2012) Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119:2579-2589.
- Amengual JE, Johannet P, Lombardo M, Zullo K, Hoehn D, et al. (2015) Dual Targeting of Protein Degradation Pathways with the Selective HDAC6 Inhibitor ACY-1215 and Bortezomib Is Synergistic in Lymphoma. Clinical cancer research: an official journal of the American Association for Cancer Research21:4663-4675.
- Guerrant W, Patil V, Canzoneri JC, Oyelere AK (2012) Dual targeting of histone deacetylase and topoisomerase II with novel bifunctional inhibitors. Journal of medicinal chemistry55:1465-1477.
- Guerrant W, Patil V, Canzoneri JC, Yao LP, Hood R, et al. (2013) Dual-acting histone deacetylase-topoisomerase I inhibitors. Bioorg Med ChemLett 23:3283-3287.
- Chen GL, Wang LH, Wang J, Chen K, Zhao M, et al. (2013) Discovery of a small molecular compound simultaneously targeting RXR and HADC: design, synthesis, molecular docking and bioassay.Bioorg Med ChemLett 23:3891-3895.
- Chen JB, Chern TR, Wei TT, Chen CC, Lin JH, et al. (2013) Design and synthesis of dual-action inhibitors targeting histone deacetylases and 3-hydroxy-3-methylglutaryl coenzyme A reductase for cancer treatment. Journal of medicinal chemistry 56:3645-3655.
- Tavallai S, Hamed HA, Grant S, Poklepovic A, Dent P (2014)Pazopanib and HDAC inhibitors interact to kill sarcoma cells. Cancer biology & therapy 15:578-585.
- Hanke NT, Garland LL, Baker AF (2015)Carfilzomib combined with suberanilohydroxamic acid (SAHA) synergistically promotes endoplasmic reticulum stress in non-small cell lung cancer cell lines. J Cancer Res ClinOncol.
- Wang G, Niu X, Zhang W, Caldwell JT, Edwards H, et al. (2015) Synergistic antitumor interactions between MK-1775 and panobinostat in preclinical models of pancreatic cancer. Cancer letters356:656-668.
- Govindaraj C, Tan P, Walker P, Wei A, Spencer A, et al. (2014) Reducing TNF receptor 2+ regulatory T cells via the combined action of azacitidine and the HDAC inhibitor, panobinostat for clinical benefit in acute myeloid leukemia patients. Clinical cancer research: an official journal of the American Association for Cancer Research 20:724-735.
- Xiao K, Li YP, Wang C, Ahmad S, Vu M, et al. (2015) Disulfide cross-linked micelles of novel HDAC inhibitor thailandepsin A for the treatment of breast cancer. Biomaterials 67: 183-193.
- Yao Y, Tu Z, Liao C, Wang Z, Li S, et al. (2015) Discovery of Novel Class I Histone Deacetylase Inhibitors with Promising in vitroand in vivoAntitumor Activities. Journal of medicinal chemistry 58:7672-7680.
- Chen CH, Lee CH, Liou JP, Teng CM, Pan SL (2015) Molecular mechanisms underlying the antitumor activity of (E)-N-hydroxy-3-(1-(4-methoxyphenylsulfonyl)-1,2,3,4-tetrahydroquinolin-6-yl)acry lamide in human colorectal cancer cells in vitro and in vivo. Oncotarget 6:35991-6002.
- Kong HS, Tian S, Kong Y, Du G, Zhang L, et al. (2012) Preclinical studies of YK-4-272, an inhibitor of class II histone deacetylases by disruption of nucleocytoplasmic shuttling. Pharm Res 29:3373-3383.
- Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, et al. (2013) Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One 8:e68915.
- Matthews GM, Mehdipour P, Cluse LA, Falkenberg KJ, Wang E, et al. (2015) Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood126:2392-2403.
- Suzuki T, Kasuya Y, Itoh Y, Ota Y, Zhan P, et al. (2013) Identification of highly selective and potent histone deacetylase 3 inhibitors using click chemistry-based combinatorial fragment assembly. PLoS One 8:e68669.
- Chan CT, Qi J, Smith W, Paranol R, Mazitschek R, et al. (2014) Syntheses and discovery of a novel class of cinnamichydroxamates as histone deacetylase inhibitors by multimodality molecular imaging in living subjects. Cancer Res 74:7475-7486.
- Seidel C, Schnekenburger M, Mazumder A, Teiten MH, Kirsch G, et al. (2015) 4-Hydroxybenzoic acid derivatives as HDAC6-specific inhibitors modulating microtubular structure and HSP90alpha chaperone activity against prostate cancer. BiochemPharmacol.
- Yang Z, Wang T, Wang F, Niu T, Liu Z, et al. (2015) Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. Journal of medicinal chemistry.
- Suzuki T, Ota Y, Ri M, Bando M, Gotoh A, et al. (2012) Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. Journal of medicinal chemistry 55:9562-9575.
- Lopez G, Bill KL, Bid HK, Braggio D, Constantino D, et al. (2015) HDAC8, A Potential Therapeutic Target for the Treatment of Malignant Peripheral Nerve Sheath Tumors (MPNST). PLoS One 10:e0133302.
- Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, et al. (2015) Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis 6:e1657.
- Wilmott JS, Colebatch AJ, Kakavand H, Shang P, Carlino MS, et al. (2015) Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod Pathol 28:884-894.
- Chakrabarti A, Oehme I, Witt O, Oliveira G, Sippl W, et al. (2015) HDAC8: a multifaceted target for therapeutic interventions. Trends PharmacolSci 36:481-492.
- Wagner FF, Zhang YL, Fass DM, Joseph N, Gale JP, et al. (2015) Kinetically Selective Inhibitors of Histone Deacetylase 2 (HDAC2) as Cognition Enhancers. Chemical science 6:804-815.
- Newbold A, Matthews GM, Bots M, Cluse LA, Clarke CJ, et al. (2013) Molecular and biologic analysis of histone deacetylase inhibitors with diverse specificities. Mol Cancer Ther12:2709-2721.
- Stubbs MC, Kim W, Bariteau M, Davis T, Vempati S, et al. (2015) Selective Inhibition of HDAC1 and HDAC2 as a Potential Therapeutic Option for B-ALL. Clinical cancer research: an official journal of the American Association for Cancer Research 21:2348-2358.
- Valente S, Trisciuoglio D, De Luca T, Nebbioso A, Labella D, et al. (2014) 1,3,4-Oxadiazole-containing histone deacetylase inhibitors: anticancer activities in cancer cells. Journal of medicinal chemistry 57:6259-6265.
- Kasparkova J, Kostrhunova H, Novakova O, Krikavova R, Vanco J, et al. (2015) A Photoactivatable Platinum(IV) Complex Targeting Genomic DNA and Histone Deacetylases. AngewChemInt Ed Engl.
- Qian C, Lai CJ, Bao R, Wang DG, Wang J, et al. (2012) Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clinical cancer research: an official journal of the American Association for Cancer Research 18:4104-4113.
- Wang J, Pursell NW, Samson ME, Atoyan R, Ma AW, et al. (2013) Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol Cancer Ther 12:925-936
- Zhang L, Zhang Y, Mehta A, Boufraqech M, Davis S, et al. (2015) Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 6:9073-9085.
- De Bellis F, Carafa V, Conte M, Rotili D, Petraglia F, et al. (2014) Context-selective death of acute myeloid leukemia cells triggered by the novel hybrid retinoid-HDAC inhibitor MC2392. Cancer Res 74:2328-2339.
- Tang C, Li C, Zhang S, Hu Z, Wu J, et al. (2013) Novel Bioactive Hybrid Compound Dual Targeting Estrogen Receptor and Histone Deacetylase for the Treatment of Breast Cancer. Journal of medicinal chemistry 58:4550-4572.
- Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, et al. (2015) ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Me 7:831-847.
- Huang HL, Peng CY, Lai MJ, Chen CH, Lee HY, et al. (2015) Novel oral histone deacetylase inhibitor, MPT0E028, displays potent growth-inhibitory activity against human B-cell lymphoma in vitro and in vivo. Oncotarget 6:4976-4991.
- Liu C, Ding H, Li X, Pallasch CP, Hong L, et al. (2015) A DNA/HDAC dual-targeting drug CY190602 with significantly enhanced anticancer potency. EMBO Mol Med 7:438-449.
- Hancock WW, Akimova T, Beier UH, Liu Y, Wang L (2012) HDAC inhibitor therapy in autoimmunity and transplantation. Annals of the rheumatic diseases 71:i46-i54.
- Poljak M, Lim R, Barker G, Lappas M (2014) Class I to III histone deacetylases differentially regulate inflammation-induced matrix metalloproteinase 9 expression in primary amnion cells. Reproductive sciences21:804-813.
- Halili MA, Andrews MR, Sweet MJ, Fairlie DP (2009) Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem 9:309-319.
- Royce SG, Karagiannis TC (2014) Histone deacetylases and their inhibitors: new implications for asthma and chronic respiratory conditions. Current opinion in allergy and clinical immunology 14:44-48.
- Akimova T, Beier UH, Liu Y, Wang L, Hancock WW (2012)Histone/protein deacetylases and T-cell immune responses. Blood 119:2443-2451.
- Hsieh IN, Liou JP, Lee HY, Lai MJ, Li YH et al. (2014)Preclinical anti-arthritic study and pharmacokinetic properties of a potent histone deacetylase inhibitor MPT0G009. Cell Death Dis5:e1166.
- Cantley MD, Fairlie DP, Bartold PM, Marino V, Gupta PK, et al. (2015) Inhibiting histone deacetylase 1 suppresses both inflammation and bone loss in arthritis. Rheumatology (Oxford) 54:1713-1723.
- Hawtree S, Muthana M, Wilkinson JM, Akil M, Wilson AG (2015) Histone deacetylase 1 regulates tissue destruction in rheumatoid arthritis. Hum Mol Genet 24:5367-5377.
- Cleophas MC, Crisan TO, Lemmers H, Toenhake-Dijkstra H, Fossati G, et al. (2014) Suppression of monosodium urate crystal-induced cytokine production by butyrate is mediated by the inhibition of class I histone deacetylases. Annals of the rheumatic diseases.
- Choi SY, Ryu Y, Kee HJ, Cho SN, Kim GR, et al. (2015) Tubastatin A suppresses renal fibrosis via regulation of epigenetic histone modification and Smad3-dependent fibrotic genes. VasculPharmacol72:130-140.
- Li Y, Zhao T, Liu B, Halaweish I, Mazitschek R, Duan X et al. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg 78:378-385.
- Tsuji G, Okiyama N, Villarroel VA, Katz SI (2015) Histone deacetylase 6 inhibition impairs effector CD8 T-cell functions during skin inflammation. The Journal of allergy and clinical immunology135:1228-1239.
- Li S, Fossati G, Marchetti C, Modena D, Pozzi P, et al. (2015) Specific inhibition of histone deacetylase 8 reduces gene expression and production of proinflammatory cytokines in vitro and in vivo . J BiolChem290:2368-2378.
- Di Liddo R, Valente S, Taurone S, Zwergel C, Marrocco B, et al. (2016) Histone deacetylase inhibitors restore IL-10 expression in lipopolysaccharide-induced cell inflammation and reduce IL-1beta and IL-6 production in breast silicone implant in C57BL/6J wild-type murine model. Autoimmunity 2016:1-11.
- Henry TR (2013)The history of valproate in clinical neuroscience. Psychopharmacology bulletin 37:5-16.
- Didonna A, Opal P (2015)The promise and perils of HDAC inhibitors in neurodegeneration. Annals of clinical and translational neurology2: 79-101.
- Zwergel C, Valente S, Jacob C, Mai A (2015) Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin Drug Discov 10:599-613.
- Suo H, Wang P, Tong J, Cai L, Liu J, Huang D et al. (2015) NRSF is an essential mediator for the neuroprotection of trichostatin A in the MPTP mouse model of Parkinson's disease. Neuropharmacology99: 67-78.
- Kozikowski AP, Chen Y, Gaysin A, Chen B, D'Annibale MA, et al. (2007) Functional differences in epigenetic modulators-superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. Journal of medicinal chemistry50:3054-3061.
- Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, et al. (2010) Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am ChemSoc132:10842-10846.
- Zhang L, Liu C, Wu J, Tao JJ, Sui XL, et al. (2014) Tubastatin A/ACY-1215 improves cognition in Alzheimer's disease transgenic mice. Journal of Alzheimer's disease: JAD 41 1193-1205.
- Zhang L, Sheng S, Qin C (2013)The role of HDAC6 in Alzheimer's disease. Journal of Alzheimer's disease: JAD 33:283-295.
- Schroeder FA, Lewis MC, Fass DM, Wagner FF, Zhang YL, et al. (2013) A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS One8:e71323.
- Benito E, Urbanke H, Ramachandran B, Barth J, Halder R, et al. (2015) HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Invest 125:3572-3584.
- Mielcarek M, Zielonka D, Carnemolla A, Marcinkowski JT, et al. (2015) HDAC4 as a potential therapeutic target in neurodegenerative diseases: a summary of recent achievements. Front Cell Neurosci 9:42.
- Burli RW, Luckhurst CA, Aziz O, Matthews KL, Yates D, et al. (2013) Design, synthesis, and biological evaluation of potent and selective class IIa histone deacetylase (HDAC) inhibitors as a potential therapy for Huntington's disease. Journal of medicinal chemistry56:9934-9954.
- Choong CJ, Sasaki T, Hayakawa H, Yasuda T, Baba K, et al. (2015) A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson's disease. Neurobiol Aging
- Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL (2010) Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 3:2751-67
- Rasmussen TA, Schmeltz SO, Brinkmann C, Wightman F, Lewin SR, et al. (2013) Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Human vaccines&immunotherapeutics 9:993-1001.
- Badia R, Grau J, Riveira-Munoz E, Ballana E, Giannini G et al. (2015) The thioacetate-omega(gamma-lactam carboxamide) HDAC inhibitor ST7612AA1 as HIV-1 latency reactivation agent. Antiviral Res123:62-69.
- Palmisano I, Della Chiara G, D'Ambrosio RL, Huichalaf C, Brambilla P, et al. (2012) Amino acid starvation induces reactivation of silenced transgenes and latent HIV-1 provirus via down-regulation of histone deacetylase 4 (HDAC4). Proceedings of the National Academy of Sciences of the United States of America109:E2284-E2293.
- Husain M, Cheung CY (2014) Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. Journal of virology 88:11229-11239.
- Ghosh SK, Perrine SP, Williams RM, Faller DV (2012) Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside antiviral agents. Blood119:1008-1017.
- Zhou L, He X, Gao B, Xiong S (2015) Inhibition of Histone Deacetylase Activity Aggravates Coxsackievirus B3-Induced Myocarditis by Promoting Viral Replication and Myocardial Apoptosis. Journal of virology89:10512-10523.
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, et al. (2011)Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol29:255-265.

