Keywords
Cholangitis, Sclerosing; Immunoglobulin E; Immunoglobulin G; Pancreatitis, Chronic; Retroperitoneal Fibrosis
INTRODUCTION
Currently, a novel clinicopathological entity of IgG4-related sclerosing disease has been proposed [1, 2, 3, 4]. The entity was first recognized as an extra-pancreatic complication of a recently proposed unique type of chronic pancreatitis, now known as autoimmune pancreatitis, in which autoimmune mechanisms are supposedly to be involved [5, 6, 7, 8]. IgG4-related sclerosing disease is classified as a systemic disease, including autoimmune pancreatitis itself, and is characterized by high serum IgG4 concentrations and extensive infiltration of IgG4-positive plasma cells into a variety of organs, such as the bile duct [9], salivary gland [10], retroperitoneum [11], kidney [12], lung [13] and prostate [14]. Another distinguishing clinical feature of IgG4-related sclerosing disease is that it responds well to steroid therapy. With the increased recognition of this clinical entity, an increased number of cases with IgG4-related retroperitoneal fibrosis have been reported as part of autoimmune pancreatitis [11, 15, 16, 17]; whereas, IgG4-related retroperitoneal fibrosis has been reported to develop independently of autoimmune pancreatitis relatively frequently [18]. However, because of its rarity and the lack of standardized diagnostic criteria for IgG4-related retroperitoneal disease, it is not always easy to make a differential diagnosis of those cases lacking clinical manifestations related to autoimmune pancreatitis. In addition, the long-term natural course of the disease remains unknown.
We describe herein a case of the metachronous recurrence of IgG4-related retroperitoneal fibrosis with overt bilateral hydronephrosis after a 5-year history of spontaneous remission. The primary clinical presentation was obstructive jaundice, presumably caused by IgG4-related sclerosing cholangitis, which also clinically resolved itself, but persisted insidiously for a long time without autoimmune pancreatitis features.
CASE REPORT
In October 2003, an 80-year-old Japanese man with a 2-week history of appetite loss was admitted to our hospital because of generalized icterus and marked elevation of liver enzymes. On admission, he was 162 cm tall and weighed 47.8 kg, blood pressure was 130/78 mmHg and temperature was 36.7°C. There was no medical history of alcohol abuse. Physical examination revealed no abnormal findings, except for jaundice of the conjunctiva and skin. Laboratory findings on the first admission are shown in Table 1. Elevation of liver enzymes with hyperbilirubinemia was demonstrated. Serological tests for hepatitis A, B and C were negative. The levels of tumor markers; CA 19-9 and CEA, were within the normal range. The serum concentration of IgG was elevated to 2,240 mg/dL (reference range: 870-1,700 mg/dL), whereas antinuclear antibody was negative. Upon abdominal computed tomography (CT), intrahepatic bile duct dilatation upstream of the hepatic hilum was shown; however, the lesion responsible for causing obstruction was not visualized (Figure 1a). No abnormalities in the pancreas were identified, whereas a series of soft tissue masses having good enhancement effects surrounding the abdominal aorta and common iliac artery were incidentally observed (Figure 1b). On magnetic resonance imaging (MRI), the mass was found to be diffusely hypointense on T1-weighted imaging and to be well-enhanced, which suggested inflammatory fibrous tissue, that is, retroperitoneal fibrosis. Percutaneous transhepatic biliary drainage was performed and subsequent cholangiography via a percutaneous transhepatic biliary drainage tube showed a cancerous stricture of the biliary tract at the hepatic hilum (Figure 2). The initial diagnosis was obstructive jaundice caused by cholangiocarcinoma simultaneously complicated with retroperitoneal fibrosis; however, unexpectedly, the jaundice and elevation of liver enzymes improved spontaneously. The patient was discharged without recurrence and visited the hospital for regular biochemistry checks and imaging thereafter.
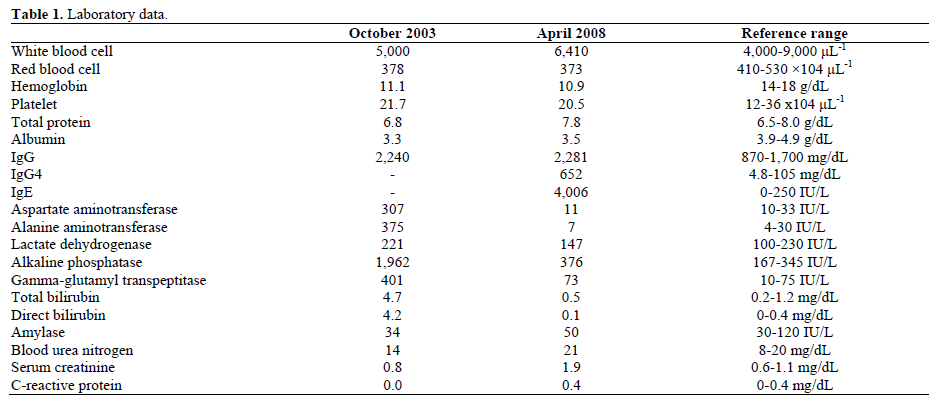
Figure 1. Abdominal enhanced CT imaging with contrast medium in
October 2003. a. Intra-hepatic bile duct dilatations were seen;
however, the lesion responsible which impaired biliary flow was not
identified. A Soft tissue mass having good enhancement surrounding
the abdominal aorta and common iliac artery was incidentally
demonstrated. b. There were no signs of hydronephrosis suggesting
encasement of the ureter.
Figure 2. Cholangiography via a percutaneous transhepatic biliary
drainage tube showed a cancerous stricture at the hepatic duct just
below the junction of the bilateral biliary ducts. The initial diagnosis
was obstructive jaundice caused by cholangiocarcinoma arising from
a hepatic hilum.
The high serum IgG concentrations persisted and the serum IgG4 concentration in June 2004 was found to be elevated to 576 mg/dL (reference range: 4.8-105 mg/dL), which led us to consider the possible association with autoimmune pancreatitis, although there were no clinical symptoms and imaging confirming autoimmune pancreatitis. Regression of both intra-hepatic biliary dilatation (Figure 3a) and retroperitoneal fibrosis (Figure 3b) was confirmed on abdominal CT in December 2005.
Figure 3. Abdominal enhanced CT imaging with contrast medium in
December 2005. Intra-hepatic bile duct dilatations disappeared (a.)
and retroperitoneal fibrosis also resolved spontaneously (b.).
The patient remained in good condition until late April 2008, when he complained of decreased urinary excretion and the level of serum creatinine was found to be elevated to 1.9 mg/dL (reference range: 0.6-1.1 mg/dL). Abdominal CT disclosed bilateral hydronephrosis (Figure 4a) caused by retroperitoneal re-thickening together with encasement of both ureters (Figure 4b). The patient was readmitted to the hospital to evaluate hydronephrosis owing to possible recurrence of retroperitoneal fibrosis in May 2008. On admission, he was 162 cm tall and weighed 48.4 kg, blood pressure was 156/64 mmHg and temperature was 36.6°C. Physical examination revealed no abnormality. Laboratory findings before the second admission are shown in Table 1. Liver dysfunction was absent; however, abnormal renal function was shown. Serum levels of IgG and IgG4 were elevated to 2,281 and 652 mg/dL, respectively. On MRI imaging, the nature of the retroperitoneal mass was found to be identical to that seen 5 years previously. There were no abnormalities suggesting malignancy in the lung, stomach and colon. Upon ERCP, the biliary stricture at the hepatic hilum observed on first admission was still present, although to a lesser extent, whereas the main pancreatic duct was normal without narrowing, suggesting that sclerosing cholangitis had persisted for a long time in a subclinical fashion (Figure 5). Gradual, but significant, elevation of the serum IgG4 concentration (September 2007: 389 mg/dL; November 2007: 407 mg/dL; April 2008: 652 mg/dL; Figure 6) resulting in overt mechanical obstruction of ureters, led us to consider possible IgG4-related sclerosing disease. Considering his advanced age (85 years old), the risk accompanying the procedure and the low overall estimated risk of malignancy as seen in a general check-up, we avoided carrying out a biopsy of the periaortic lesion. Subsequent to oral prednisolone introduction at an initial daily dose of 25 mg in June 2008, a prompt decrease in IgG as well as IgG4 levels was achieved. In addition, serum creatinine levels returned to within normal limits in parallel with the regression of retroperitoneal fibrosis. A follow-up abdominal CT disclosed that the hydronephrosis as well as the retroperitoneal thickening had improved; however, as the levels of IgG4 had slightly re-increased when the daily prednisolone dose was reduced to 5 mg, careful surveillance for recurrence was required. The serum level of IgE before the initiation of steroid therapy was found to be markedly elevated to 4,006 IU/mL (reference range: 0-250 IU/mL), which currently, one year after prednisolone introduction, still remains high at 1,194 IU/mL. However, the patient did not have any medical history and symptom suggestive of allergic constitution.
Figure 4. Abdominal CT in May 2008 before the second admission
disclosed bilateral hydronephrosis (a.) caused by sequential
thickening of the retroperitoneum with encasement of both ureters
(b.), suggesting recurrence of retroperitoneal fibrosis.
Figure 5. ERCP showed intrahepatic bile duct dilatation and the
persistence of the biliary stricture at the hepatic hilum suggestive of
sclerosing cholangitis which was observed on the first admission,
whereas the main pancreatic duct exhibited a normal appearance
without irregularity and narrowing.
Figure 6. IgG and IgG4 levels before and after the initiation of
steroid therapy. Both levels of IgG and IgG4 were significantly
elevated before the initiation of prednisolone (PSL) and subsequently
decreased to the normal range in parallel with clinical improvement.
However, the levels of IgG and igG4 increased slightly when the
daily prednisolone dose was reduced to 5 mg.
DISCUSSION
This case report provides important clinical information when dealing with patients with a fibrotic disease of unknown etiology. First, idiopathic retroperitoneal fibrosis can occur as part of a generalized disorder of a new clinical entity; IgG4-related sclerosing disease. Second, IgG4-related sclerosing disease can develop independently of autoimmune pancreatitis and on such occasions, a diagnosis is not always easy unless high serum IgG4 concentrations are noted. Third, the disease may present with a variety of unexpected clinical features, as shown in this patient, namely, spontaneous remission or sudden recurrence of retroperitoneal fibrosis after an interval of several years and the subclinical persistence of sclerosing cholangitis. In addition, unexpectedly, the level of serum IgE at retroperitoneal fibrosis recurrence was found to be markedly high, and currently it is still high even after prednisolone introduction.
Recently, reports on retroperitoneal fibrosis associated with autoimmune pancreatitis have been increasing [11, 15, 16, 17]. Autoimmune pancreatitis is a chronic inflammatory condition of the pancreas in which an autoimmune mechanism is supposed to be involved and is now recognized as a distinct clinical entity worldwide [5, 6, 7, 8]. As awareness of autoimmune pancreatitis is increasing and more cases of autoimmune pancreatitis are clinically investigated, high serum IgG4 concentrations have been recognized as its distinguishing characteristics and as a marker of autoimmune pancreatitis as well as disease activity [19]. Moreover, as the presence of various extra-pancreatic lesions related to IgG4-positive plasma cell infiltration has become evident [9, 10, 11], autoimmune pancreatitis has been proposed as a clinical aspect of generalized autoimmune IgG4-related sclerosing disease [1, 2, 3, 4].
Retroperitoneal fibrosis is an uncommon fibrotic condition of the retroperitoneum usually forming a fibro-inflammatory mass lesion around the periaortic space sometimes resulting in an obstructive uropathy [20]. The majority of etiological causes of retroperitoneal fibrosis were reported to be idiopathic, in which the disease was called chronic periaortitis, inflammatory abdominal aortic aneurysms and perianeurysmal retroperitoneal fibrosis, and it engendered the conceptual confusion [20]. However, currently, idiopathic retroperitoneal fibrosis has been defined as a non-aneurysmal form of periaortitis. Similar conditions may occur secondary to a wide spectrum of causes, including malignancies, infections and medications [20]; therefore, it is usually difficult to make a differential diagnosis and histopathology is thought to be mandatory to elucidate its etiological causes. However, in the present case, histopathological confirmation was not attempted because of the concern that biopsy of the periaortic lesion of an 85-year-old patient was risky. Steroid therapy was decided upon because of the second exacerbation of a periaortic soft tissue mass in parallel with a gradual increase in serum IgG4 concentrations, resulting in overt mechanical bilateral ureter complications. In addition, as the likelihood of spontaneous recovery seemed poor, immediate preservation of renal function in a reversible condition was thought to be required. Moreover, a previous report described by Zen et al. demonstrated that serum IgG4 levels could be predictive markers for determining the corticosteroid sensitivity of retroperitoneal fibrosis [21]. Subsequent to steroid therapy, both biochemical and morphological improvement was achieved, showing an excellent effect on the decrease in IgG4 levels together with regression of retroperitoneal fibrosis. Thus, the patient was diagnosed as having IgG4-related sclerosing disease.
When IgG4-related retroperitoneal fibrosis develops in association with autoimmune pancreatitis, the diagnosis is simple; otherwise, it is not always easy to diagnose the disease. Reports describing complexes of IgG4-related sclerosing disease, including retroperitoneal fibrosis, without pancreatic involvement have already been seen [15, 21]. The present case demonstrates, once again, that IgG4-related sclerosing disease can develop independently of autoimmune pancreatitis and the clue in making a diagnosis in such a situation is whether high serum IgG4 concentrations can be recognized. A report by Kamisawa et al. [16] dealing with autoimmune pancreatitis which developed metachronously 10 months after the occurrence of retroperitoneal fibrosis, suggested that autoimmune pancreatitis and its extra-pancreatic sclerosing complications do not necessarily occur simultaneously. Therefore, although the patient had been free of autoimmune pancreatitis for as long as 5 years, whether he would have developed autoimmune pancreatitis is unknown because of the introduction of steroid therapy.
The clinical course of the case was significant in demonstrating that the disease activity in both retroperitoneal fibrosis and sclerosing cholangitis resolved spontaneously, and that the former recurred suddenly after a 5-year interval, the latter persisted subclinically. The important point that should be kept in mind was that the recurrence of retroperitoneal fibrosis occurred in parallel with the elevation of serum IgG4 concentrations. As previously reported, in autoimmune pancreatitis [19] the serum levels of IgG4 can also be clinical markers for the disease activity of IgG4 related sclerosing disease. Another distinguishing clinical feature was that the serum level of IgE was significantly high when retroperitoneal fibrosis recurred (4,006 IU/mL). Recently, Kasashima et al. published an interesting report noting that the patient with IgG4-related inflammatory abdominal aortic aneurysms frequently showed an allergic condition, such as bronchial asthma, drug allergy and high serum IgE concentrations [22]. However, as the patient did not have any medical history or symptoms suggestive of allergic conditions, the clinical significance of the abnormally high levels of serum IgE remains unknown. Further observation associated with the levels of IgE in IgG4-related retroperitoneal fibrosis is required.
In conclusion, we describe herein a case, (successfully treated using steroid therapy), of metachronous recurrence of IgG4-related retroperitoneal fibrosis with overt bilateral hydronephrosis after a 5-year period of spontaneous remission. The primary clinical presentation was obstructive jaundice caused by IgG4-related sclerosing cholangitis and this was also clinically resolved spontaneously but persisted insidiously without features of autoimmune pancreatitis. Since steroid therapy has a favorable effect on IgG4-related sclerosing disease, we should consider the possibility of this clinical entity and monitor serum levels of IgG4 when encountering fibrotic diseases of unknown etiology. In addition, as concerns disease recurrence, long-term monitoring of serum IgG4 concentrations is mandatory,
Conflict of interest The authors have no potential conflicts of interest
References
- Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003; 38:982-4. [PMID 14614606]
- Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006; 6:132-7. [PMID 16327291]
- Hamano H, Arakura N, Muraki T, Ozaki Y, Kiyosawa K, Kawa S. Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol 2006; 41:1197-205. [PMID 17287899]
- Kamisawa T, Okamoto A. IgG4-related sclerosing disease. World J Gastroenterol 2008; 14:3948-55. [PMID 18609677]
- Okazaki K, Uchida K, Ohana M, Nakase H, Uose S, Inai M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000; 118:573-81. [PMID 10702209]
- Kim KP, Kim MH, Lee SS, Seo DW, Lee SK. Autoimmune pancreatitis: it may be a worldwide entity. Gastroenterology 2004; 126:1214. [PMID 15057766]
- Sutton R. Autoimmune pancreatitis--also a Western disease. Gut 2005; 54: 581-3. [PMID 15831898]
- Song Y, Liu QD, Zhou NX, Zhang WZ, Wang DJ. Diagnosis and management of autoimmune pancreatitis: experience from China. World J Gastroenterol 2008; 14:601-6. [PMID 18203294]
- Ichimura T, Kondo S, Ambo Y, Hirano S, Ohmi M, Okushiba S, et al. Primary sclerosing cholangitis associated with autoimmune pancreatitis. Hepatogastroenterology 2002; 49:1221-4. [PMID 12239909]
- Kamisawa T, Tu Y, Egawa N, Sakaki N, Inokuma S, Kamata N. Salivary gland involvement in chronic pancreatitis of various etiologies. Am J Gastroenterol 2003; 98:323-6. [PMID 12591049]
- Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet 2002; 359:1403-4. [PMID 11978339]
- Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant 2004; 19:474-6. [PMID 14736977]
- Hirano K, Kawabe T, Komatsu Y, Matsubara S, Togawa O, Arizumi T, et al. High-rate pulmonary involvement in autoimmune pancreatitis. Intern Med J 2006; 36:58-61. [PMID 16409315]
- Yoshimura Y, Takeda S, Ieki Y, Takazakura E, Koizumi H, Takagawa K. IgG4-associated prostatitis complicating autoimmune pancreatitis. Intern Med 2006; 45:897-901. [PMID 16946571]
- Tanabe T, Tsushima K, Yasuo M, Urushihata K, Hanaoka M, Koizumi T, et al. IgG4-associated multifocal systemic fibrosis complicating sclerosing sialadenitis, hypophysitis, and retroperitoneal fibrosis, but lacking pancreatic involvement. Intern Med 2006; 45:1243-7. [PMID 17139126]
- Kamisawa T, Chen PY, Tu Y, Nakajima H, Egawa N. Autoimmune pancreatitis metachronously associated with retroperitoneal fibrosis with IgG4-positive plasma cell infiltration. World J Gastroenterol 2006; 12:2955-7. [PMID 16718827]
- Kuwatani M, Kawakami H, Makiyama H, Onodera M, Matsumoto K, Karasawa G, Asaka M. Autoimmune pancreatitis with retroperitoneal fibrosis which responded to steroid therapy but was complicated with refractory renal dysfunction. Intern Med 2007; 46:1557-64. [PMID 17878642]
- Neild GH, Rodriguez-Justo M, Wall C, Connolly JO. Hyper-IgG4 disease: report and characterisation of a new disease. BMC Med 2006; 4:23. [PMID 17026742]
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344:732-8. [PMID 11236777]
- Vaglio A, Salvarani C, Buzio C. Retroperitoneal fibrosis. Lancet 2006; 367:241-51. [PMID 16427494]
- Zen Y, Sawazaki A, Miyayama S, Notsumata K, Tanaka N, Nakanuma Y. A case of retroperitoneal and mediastinal fibrosis exhibiting elevated levels of IgG4 in the absence of sclerosing pancreatitis (autoimmune pancreatitis). Hum Pathol 2006; 37:239-43. [PMID 16426926]
- Kasashima S, Zen Y, Kawashima A, Endo M, Matsumoto Y, Kasashima F. A new clinicopathological entity of IgG4-related inflammatory abdominal aortic aneurysm. J Vasc Surg 2009; 49:1264-71. [PMID 19217746]







