Mahesh Manjunath Gouda* and Yashodhar Prabhakar Bhandary
Yenepoya Research Centre, Yenepoya University, Deralakatte, Mangaluru, India
Corresponding Author:
Mahesh Manjunath Gouda
Yenepoya Research Centre, Yenepoya University, Deralakatte, Mangaluru, India
Tel: 0824-2204668
E-mail: maheshmg2012@gmail.com
Received date: June 05, 2018; Accepted date: June 27, 2018; Published date: June 29, 2018
Citation: Gouda MM, Bhandary YP (2018) IL-17A Mediated Downstream Changes During Acute Lung Injury. Biochem Mol Biol J 4:13. doi: 10.21767/2471-8084.100062
Keywords
IL-17A; p53; AECs; Apoptosis; ECM; Pulmonary fibrosis
Introduction
Respiration creates regular immune reaction in the lung to fight against the invading pathogens [1]. These factors promote the inflammatory changes in the airway, which triggers the production of alveolar macrophages, neutrophils, chemokines, growth factors and inflammatory cytokines [2]. Cytokines are the small glycoproteins, which is actively involved in the immune system of the body [3]. IL-17A is a one among those having the potential to activate various pathways in response to the alveolar injury. IL-17A involved in the hostdefensive role in various diseases [4]. IL-17A was thought to be produced by the T cells, but now it is known to be secreted by many innate cells such as natural killer cells, lymphoid tissue, dendritic cells (DC), and macrophages [5]. IL-17A is an extensively studied member of IL-17 cytokine family and its closest homolog is IL-17F [6]. It is reported that IL-17A induces TNF receptor associated factor 6 (TRAF6) dependent activation of nuclear factor kappa B (NFκB), which results in the enhanced cytokine expression in the airway [6]. It is also known to enhance the secretion of matrix metalloproteinases (MMP1 and MMP3) [6]. IL-17A mediated inflammation also promotes the expression of certain chemokines such as CXCL1, CXCL5 and CXCL12 [6]. The inflammation in the alveolar epithelial baseline could stimulate the activation of apoptosis markers such as caspase-3 results in the death of AECs and progression of severe injury to fibrosis [7,8].
IL-17A in acute lung injury
Alveolar epithelium is made of 90% type I alveolar epithelial cells (ATI), which are very delicate and more susceptible to injury [9,10]. The injury to the ATI cells forms the gaps in the alveolar epithelial barrier, which is then replaced by the type II alveolar epithelial cells (ATII) [8,10]. ATII cells are capable of bridging the gaps in the alveolar epithelial baseline, which begins the injury repair mechanism [10,11]. The airway epithelial injury induces the production of various inflammatory molecules such as macrophages, neutrophils, chemokines and cytokines (Figure 1) [12-14].
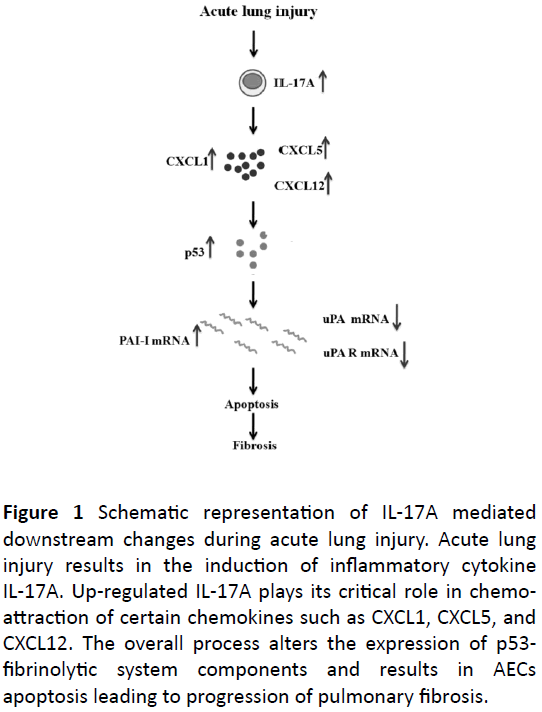
Figure 1: Schematic representation of IL-17A mediated downstream changes during acute lung injury. Acute lung injury results in the induction of inflammatory cytokine IL-17A. Up-regulated IL-17A plays its critical role in chemoattraction of certain chemokines such as CXCL1, CXCL5, and CXCL12. The overall process alters the expression of p53- fibrinolytic system components and results in AECs apoptosis leading to progression of pulmonary fibrosis.
These inflammatory molecules are involved in the production of pro-inflammatory cytokine IL-17A [15,16]. Literatures have supported the protective role of IL-17A in immunity during infection or injury of airway epithelium [8,15]. IL-17A acts on some immune cells and also on nonimmune cells such as epithelial and mesenchymal cells [15]. The elevated expression of IL-17A is known to activate the chemokines such as CXCL1, CXCL5, and CXCL12 [6]. These chemokines play a crucial role in inflammation and take part in the disease progression dependent on severity of injury [6,17]. The significant changes in the IL-17A expression are dependent on the severity of lung injury [6]. Severity of lung injury is indicated by the rate of loss of AECs at the alveolar epithelium [6]. Literatures suggest that it is possible to regulate the IL-17A mediated pro-inflammation and its associated cell death by blocking the IL-17A expression at the molecular level [18]. Inhibition of IL-17A expression could regulate the expression of certain chemokines such as CXCL1, CXCL5, CXCL12 and its mediated interlinked pathways to initiate the AEC injury healing process [8,19].
IL-17A mediated changes during acute lung injury and progression to pulmonary fibrosis
As discussed earlier, the crucial role of IL-17A is to trigger the inflammatory response against the cellular damage [6,20].
The elevated expression of IL-17A also indicates the severe progression of injury to the fibro-proliferative phase of the disease [6]. IL-17A cross-talk with p53 causes the DNA damage at the molecular level; this damage leads to breaking of bond between p53 and mdm2 [6,10]. It is also reported that p53 mediates the DNA repair mechanism. The damage beyond repair leads to the activation of apoptosis pathway by switching on the action of caspase [10,11]. Literatures also suggested that the role of IL-17A in the alteration of p53 mediated fibrinolytic system. The elevated expression of p53 in the nucleus of the AECs determines the cell function and viability [6,10].
p53 is known to regulate the fibrinolytic system components such as urokinase plasminogen activator (uPA), urokinase plasmingen activator receptor (uPAR) and urokinase plasminogen activator receptor-1 (PAI-I) [11]. The elevated expression of p53 in the nucleus promotes the production of PAI-I mRNA by suppressing uPA and uPAR mRNA production [11,19,21-23]. The impaired fibrinolytic system induces the PAI-I deposition and consequent accumulation of ECM at the injured site [11]. The severe loss of AECs at the alveolar epithelial baseline is responsible for developing the fibroproliferative phase [10]. The excessive deposition of fibroblast or ECM results in the thickening of the alveolar barrier and blocks the gaseous exchange [10,17]. These pathological changes forms the scar at the epithelial barrier, this scarring of tissue is known as fibrosis [23-25].
Discussion and Conclusion
IL-17A plays a crucial role as a pro-inflammatory cytokine during acute lung injury. The increased expression of IL-17A indicates the severity of lung injury and its development. IL-17A also involved in the mediation of several important pathways such as chemokines, p53-fibrinolytic, and apoptosis pathways. Importantly, the expression of IL-17A is key measure of lung injury progression to the fibro-proliferative phase and conversion of fibroblast to myofibroblasts. The specific interaction of IL-17A with the p53-fibrinolytic system is the major reason for impaired fibrinolytic system and development of pulmonary fibrosis. Blocking of IL-17A could serve as an effective molecular therapy to regulate the acute lung injury related health issues in society.
Acknowledgements
Authors would like to acknowledge Yenepoya Research Centre, Yenepoya University for the support.
References
- Matthay MA, Zimmerman GA (2005) Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol 33: 319-327.
- Saldiva PH, Clarke RW, Coull BA, Stearns RC, Lawrence J, et al. (2002) Lung inflammation induced by concentrated ambient air particles is related to particle composition. Am J Respir Crit Care Med 165: 1610-1617.
- Goodman RB, Pugin J, Lee JS, Matthay MA (2003) Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev 14: 523-535.
- Wilson MS, Madala SK, Ramalingam TR, Gochuico BR (2010) Bleomycin and IL-1β–mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207: 535-552.
- Onishi RM, Gaffen SL (2010) Interleukin?17 and its target genes: Mechanisms of interleukin?17 function in disease. Immunology 129: 311-321.
- Sharma J, Balakrishnan L, Datta KK, Sahasrabuddhe NA, Khan AA, et al. (2015) A knowledgebase resource for Interleukin-17 family mediated signaling. J Cell Commun Signal 9: 291-296.
- Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, et al. (2002) TGF-β1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J Immunol 168: 6470-6478.
- Gouda MM, Prabhu A, Bhandary YP (2017) Curcumin alleviates IL?17A?mediated p53?PAI?1 expression in bleomycin?induced alveolar basal epithelial cells. J Cell Biochem 119: 2222-2230.
- Fehrenbach H (2001) Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir Res 2: 33.
- Bhandary YP, Shetty SK, Marudamuthu AS, Gyetko MR, Idell S, et al. (2012) Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system. Am J Physiol Lung Cell Mol Physiol 302: L463-L473.
- Shetty S, Padijnayayveetil J, Tucker T, Stankowska D, Idell S (2008) The fibrinolytic system and the regulation of lung epithelial cell proteolysis, signaling, and cellular viability. Am J Physiol Lung Cell Mol Physiol 295: L967-L975.
- Ricard JD, Dreyfuss D, Saumon G (2001) Production of inflammatory cytokines in ventilator-induced lung injury: A reappraisal. Am J Respir Crit Care Med 163: 1176-1180.
- Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM (1991) Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J Biol Chem 266: 9912-9918.
- Yan B, Chen F, Xu L, Xing J, Wang X (2017) HMGB1-TLR4-IL23-IL17A axis promotes paraquat-induced acute lung injury by mediating neutrophil infiltration in mice. Sci Rep 7: 597.
- Wei J, Chen D (2013) IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect 60: 1-5.
- https://grantome.com/grant/NIH/K08-HL111208-02.
- Tsai HC, Velichko S, Hung LY, Wu R (2013) IL-17A and Th17 cells in lung inflammation: An update on the role of Th17 cell differentiation and IL-17R signaling in host defense against infection. Clin Dev Immunol 17: 267971.
- Li Q, Gu Y, Tu Q, Wang K, Gu X, et al. (2016) Blockade of interleukin?17 restrains the development of acute lung injury. Scand J Immunol 83: 203-211.
- https://www.ncbi.nlm.nih.gov/pubmed/29775223.
- Righetti R, Dos Santos TM, Do Nascimento Camargo L, Barbosa LR, Fukuzaki S, et al. (2017) Effects of anti-IL17 on acute lung injury induced by LPS in mice. Eur Respir J 50: PA346.
- Chow LN, Schreiner P, Ng BY, Lo B, Hughes MR, et al. (2016) Impact of a CXCL12/CXCR4 antagonist in bleomycin (BLM) induced pulmonary fibrosis and carbon tetrachloride (CCL4) induced hepatic fibrosis in mice. Plos one 11: e0151765.
- Bhandary YP, Shetty SK, Marudamuthu AS, Ji HL, Neuenschwander PF et al. (2013) Regulation of lung injury and fibrosis by p53-mediated changes in urokinase and plasminogen activator inhibitor-1. Am J Pathol 183: 131-143.
- Tucker TA, Jeffers A, Alvarez A, Owens S, Koenig K, et al. (2014) Plasminogen activator inhibitor-1 deficiency augments visceral mesothelial organization, intrapleural coagulation, and lung restriction in mice with carbon black/bleomycin–induced pleural injury. Am J Respir Cell Mol Biol 50: 316-327.
- Shi K, Jiang J, Ma T, Xie J, Duan L, et al. (2014) Pathogenesis pathways of idiopathic pulmonary fibrosis in bleomycin-induced lung injury model in mice. Respir Physiol Neurobiol 190: 113-117.
- Marudamuthu AS, Shetty SK, Bhandary YP, Karandashova S, Thompson M, et al. Plasminogen activator inhibitor-1 suppresses profibrotic responses in fibroblasts from fibrotic lungs. J Biol Chem 290: 9428-9441.

