Keywords
Ceramides; Interleukin-1; Nitric Oxide Synthase Type II; Sphingomyelin Phosphodiesterase; Insulin-Secreting Cells
Abbreviations
CER: ceramide; DES: desipramine; FB: fumonisin B1; IL-1beta: interleukin 1beta; iNOS: inducible nitric oxide synthetase; MSM: 3-O-methylsphingomyelin; MTT: 3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide; NO: nitric oxide; PTDC: pyrrolidine dithiocarbamic acid; SMases: sphingomyelinases
INTRODUCTION
It is well established that cytokine (e.g., IL-1beta)-mediated metabolic dysfunction and demise of the islet beta cell is attributable, at least to a large degree, to the increase in the expression of iNOS and subsequent generation of NO. Potential cytotoxic effects of intracellularly-generated NO are demons-trable in variety of insulin-secreting cells, including human islets, rat islets, mouse islets and clonal beta cell preparations [1, 2, 3, 4, 5]. While there is an ongoing debate on whether cytokine-induced death of the islet beta cell involves apoptotic, necrotic or other processes, it is well accepted that IL-1beta-induced increase in the intracellular NO levels exerts direct effects on various cellular compartments, including the mitochondrion and the nucleus [6, 7].
Recent data from our laboratory demonstrated IL-1beta-mediated effects on islet mitochondria, including loss in mitochondrial membrane potential, leakage of mitochondrial cytochrome c into the cytosolic compartment, leading to the activation of specific caspases and terminating in cell death [6]. We also observed similar changes in the mitochondria in isolated beta cells incubated in the presence of exogenously added biologically active, cell-permeable analogs of ceramide (CER) (C2-CER), but not its inactive analog, dihydro-CER [6]. Together, our findings implicated that CER- or IL-1beta-mediated effects on isolated beta cells might involve common signaling pathways leading to mitochondrial damage.
To further evaluate if IL-1beta-induced metabolic dysfunction of the islet beta cell requires intracellular CER, we undertook the current study to examine potential effects of known pharmacological inhibitors of CER biosynthesis on IL-1beta-induced iNOS expression, NO release, and loss in metabolic function in INS 832/13 cells. We utilized known inhibitors of neutral as well as acidic sphingomyelinases (SMases) or a known inhibitor of CER synthase to address this question. Our findings suggest that specific indices of IL-1beta-mediated effects on the islet beta cell, namely iNOS expression, NO release, and loss in cell viability are resistant to these inhibitors, suggesting that such signaling steps may not be critical for IL-1beta-induced metabolic dysfunction of the beta cell.
MATERIALS AND METHODS
IL-1beta was purchased from R&D Systems (Minneapolis, MN, USA), Griess reagent, desipramine, fumonisin B1, and pyrrolidine dithiocarbamic acid (PTDC) was purchased from Sigma (St Louis, MO, USA). 3-O-methylsphingomyelin (MSM) was purchased from Biomol Research Laboratories (Plymouth Meeting, PA, USA). Affinity purified, monoclonal antisera directed against iNOS and mouse monoclonal alpha-tubulin antibody were purchased from Transduction Laboratories (Lexington, KY, USA) and Calbiochem (La Jolla, CA, USA), respectively. Cell viability assay kit and cell death detection ELISA PLUS for the in vitro determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) during cell death were purchased from Roche (Indianapolis, IN, USA)
Cell Culture
INS 832/13 cells were kindly provided by Dr. Chris Newgard (Duke University Medical Center, Durham, NC, USA), and were cultured in RPMI 1640 medium supplemented with 1 mmol/L sodium pyruvate, 50 μmol/L 2-mercaptoethanol and 10 mmol/L HEPES at pH 7.4. The medium was changed twice weekly, and cells were trypsinized and ubcloned weekly [6, 7, 8].
Quantitation of IL-1beta-Induced Nitrite Release
INS 832/13 cells grown in 24-well plates were treated with PTDC, a known inhibitor of NF-kappaB, for 2 h prior to the addition of IL-1beta. Cells were also treated with inhibitors of CER biosynthesis, namely desipramine (DES; 10 μmol/L), methylsphingomyelin (MSM; 10 μmol/L) or fumonisin B1 (FB; 10 μmol/L) half-hour prior to the addition of IL-1beta (600 pmol/L; 24 h). The medium was collected after 24 h and centrifuged at 100 g for 5 min. Equal volumes of medium and Griess reagent were mixed, and the absorbance was measured at 540 nm as we previously described [6, 7, 8].
Quantitation of IL-1beta-Induced iNOS Expression
INS 832/13 cells grown in 24-well plates were treated with PTDC, an inhibitor of NF-kappaB, (2 h prior to the addition of IL-1beta), or inhibitors of CER biosynthesis, namely DES, MSM or FB half-hour prior to the addition of IL-1beta (600 pmol/L; 24 h). Extracted proteins (40 μg) from different conditions were separated by SDS-PAGE and the resolved proteins were transferred to a nitrocellulose membrane by wet transfer as we previously described [7, 8]. Blots were then probed with either anti-iNOS or alpha-tubulin antibodies. Immune-complexes were detected using Enhanced Chemiluminescent kit (Amersham Biosciences, Piscataway, NJ, USA).
Determination of Metabolic Cell Viability
INS 832/13 cells, seeded at a density of 1x106 cells/mL in round-bottomed 96-well plates, were treated with diluent alone or MSM as indicated in the text half-hour prior to the addition of IL-1beta (600 pmol/L; 24 h). Cell viability was assessed using a colorimetric assay (at 550-690 nm) using 3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide (MTT), which measures the reduction of MTT into the blue formazan product by metabolically active cells.
Determination of DNA Fragmentation by ELISA
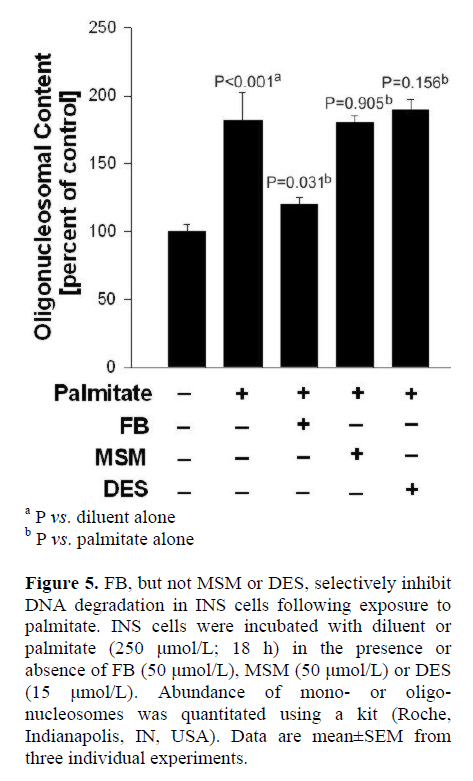
INS 832/13 cells were incubated with diluent or palmitate (250 μmol/L; 18 h) in the presence of diluent alone or FB (50 μmol/L), MSM (50 μmol/L), or DES (15 μmol/L). Abundance of mono- or oligo-nucleosomes was quantitated using an ELISA kit. The assay is based on the quantitative “sandwich enzyme immunoassay” principle using mouse monoclonal antibodies directed against DNA and histones. This allows the specific determination of mono- and oligo-nucleosomes in the cytoplasmic fraction of cell lysates. The samples placed in a streptavidin-coated microplate are incubated with a mixture of anti-histone-biotin and anti-DNA-peroxidase antibodies. During the incubation period, nucleosomes are captured by anti-histone-biotin antibody, whereas, anti-DNA-peroxidase binds to the DNA part of the nucleosomes. After removal of the unbound antibodies, the amount of peroxidase retained in the immune complex is photometrically determined with 2,2'-azino-bis (3-ethylbenzthiazoline-6)-sulfonic acid (ABTS) as the substrate.
Protein Assay
Protein concentrations were determined by dye-binding method of Bradford using bovine serum albumin as the standard.
STATISTICS
Data are reported as mean values together with SEM or SD. Statistical significance of differences between the control and experimental groups was determined by ANOVA and P values less than 0.05 were considered significant. The Sigma StatTM, statistical software, Version 1, was used to analyze the data.
RESULTS
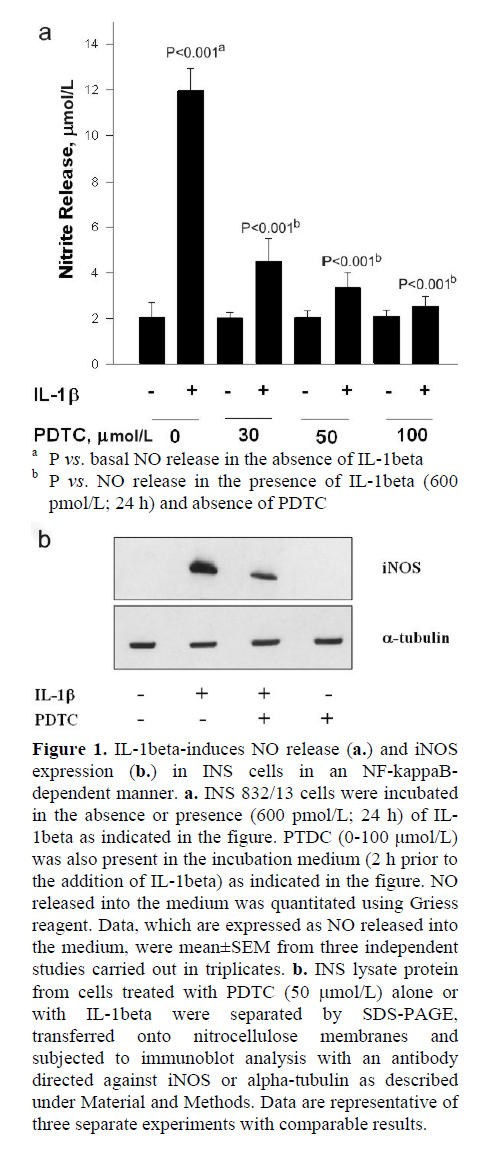
Initial experiments were aimed at verifying our model system that we employed in the present study by quantitating IL-1beta-induced iNOS expression and NO release (Figure 1a); a robust increase in NO release in INS 832/13 cells incubated with IL-1beta was demonstrated (P<0.001). Also, IL-1beta-induced NO release was markedly (P<0.001) inhibited by PTDC, a known inhibitor of NF-kappaB activation [9]. We observed greater than 60% inhibition in IL-1beta-induced NO release by PTDC at a concentration as low as 30 μmol/L. Compatible with these data are our findings in Figure 1b, which demonstrate a marked inhibition in IL-1beta-induced iNOS expression by PTDC. Together, these data demonstrate that IL-1beta-induces iNOS expression and NO release in an NF-kappaB-dependent manner. In the subsequent experiments performed in the present study, we utilized IL-1beta-induced iNOS expression and NO release as indices to examine potential roles for SMase or CER synthase activation, if any, on IL-1beta-mediated effects on INS 832/13 cells.
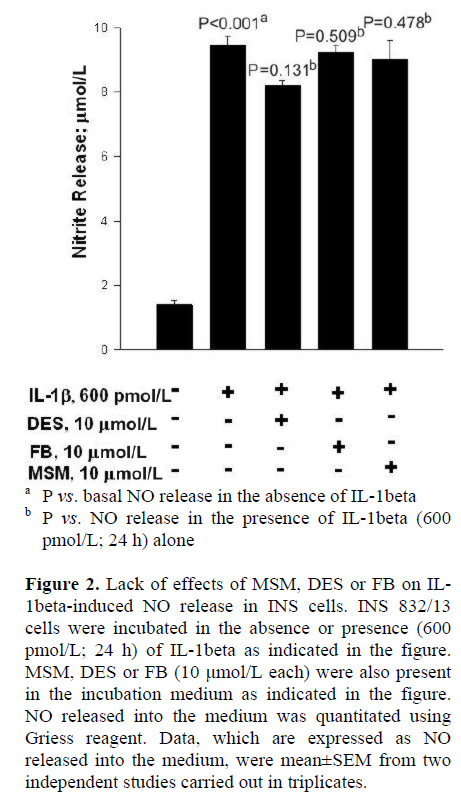
At the outset, we examined effects of two-structurally dissimilar inhibitors of SMases, namely MSM [10, 11, 12, 13], or DES [14, 15] on IL-1beta-induced NO release from INS 832/13 cells. Data in Figure 2 indicate that incubation of these cells with IL-1beta (600 pmol/L; 24 h) significantly (nearly five-fold) increased NO release compared to untreated control cells (P<0.001). However, we observed no effects of either DES or MSM (10 μmol/L each) on IL-1beta-induced NO release in these cells (P=0.131 and P=0.478, respectively; Figure 2). At this concentrations, both DES and MSM have been shown to inhibit the target acidic or neutral SMase activities [10, 11, 12, 13, 14, 15]. In addition, to further rule out the possibility that IL-1beta mediates its effects via the activation of CER synthase, we studied IL-1beta-induced NO release in these cells in the presence of FB (10 μmol/L), a known inhibitor of CER synthase [16, 17, 18, 19]. Data in Figure 2 indicate no demonstrable effects of FB on IL-1beta-induced NO release (P=0.509). Compatible with data in Figure 2 are our findings in Figure 3, which indicate no significant effects of either MSM or FB on IL-1beta-induced iNOS expression. In summary, these data implicate involvement of neither SMases nor CER synthase in IL-1beta-mediated effects on INS 832/13 cells.
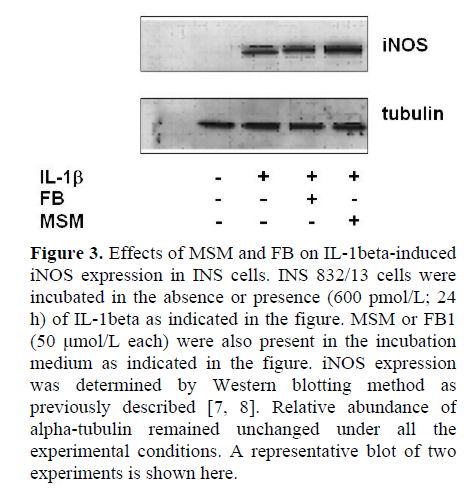
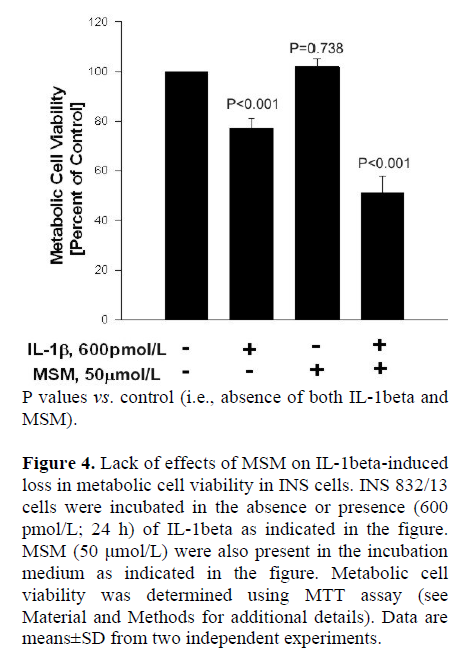
Next, we utilized these SMase or CER synthase inhibitors to further determine if these signaling steps play regulatory roles in IL-1beta-mediated loss in the metabolic cell viability of the beta cell, which was quantitated by MTT assay (see Materials and Methods for additional details). Data in Figure 4 indicate significant reduction in metabolic cell viability of INS 832/13 cells following exposure to IL-1beta (P<0.001). MSM (even up to a concentration of 50 μmol/L), by itself, exerted no effects on the metabolic cell viability of these cells in the absence of IL-1beta (P=0.738). Furthermore, it failed to protect these cells against IL-1beta-induced loss in cell viability (P<0.001). We also observed no significant protective effects of either DES or FB on IL-1beta-induced loss in the metabolic cell viability in these cells under our current experimental conditions (additional data not shown). Together, these data indicate that pharmacological inhibitors of SMases or CER synthase (Figures 2, 3, and 4) exert no protective effects against IL-1beta-mediated iNOS expression, NO release and loss in metabolic cell viability, further supporting our hypothesis that these signaling steps may not be necessary for the cytotoxic effects of IL-1beta.
In the last series of studies, we verified if FB (employed in the above studies) affords protection against palmitate-induced dysfunction in INS 832/13 cells. This was determined by quantitation of oligonucleosomal content, an index of DNA degradation, in cells incubated in the presence of palmitate and in the absence or presence of FB. Data in Figure 5 demonstrate a significant increase in the oligonucleosomal content in cells incubated with palmitate (P<0.001), which was inhibited significantly by FB (P=0.031). It should be noted that neither MSM nor DES exerted any protective effects against palmitate-induced DNA degradation in these cells (P=0.905, and P=0.156, respectively), further supporting our model with respect to specificity of the inhibitors used in the current investigation. Taken together, our findings indicate that neither the SMase activation nor CER synthase signaling steps are required for IL-1beta-induced iNOS expression, NO release and loss in metabolic cell viability in insulin-secreting INS 832/13 cells.
DISCUSSION
One of the specific objectives of this study was to determine if intracellular generation of CER (i.e., either via SMase activation or via the de novo biosynthetic pathway) could contribute toward IL-1beta-mediated effects on iNOS expression and NO release in isolated beta cells. We examined such a possibility through the use of two structurally-distinct, known inhibitors of SMases and a known inhibitor of CER synthase. Our results appear to indicate that neither of these signaling steps are necessary for IL-1beta-induced effects on iNOS expression and subsequent generation of cytotoxic NO release. To our knowledge, this represents the first study that directly examined contributory roles for SMase activation in IL-1beta-mediated effects on the beta cell using known pharmacological inhibitors of these enzymes.
Several lines of evidence tend to support a critical regulatory role for CER in the metabolic dysfunction of the islet beta cell [20]. Initial studies from Sjoholm’s laboratory demonstrated inhibitory effects of cell-permeable analogs of CER on insulin production and mitogenesis in the beta cell [21]. They also reported that CER mimicked the effects of IL-1beta under such experimental conditions. Follow-up studies by these investigators demonstrated that IL-1beta-mediated beta cell dysfunction is due to the generation of intracellular NO, CERs and prostaglandins in insulin-secreting RIN cells [22]. Compatible with these data, Welsh [23] reported that IL-1beta-induced CER and diacylglycerol generation leads to activation of c-Jun kinase and ATF-2 in insulin-secreting RIN cells.
While the above studies appear to support the postulation that IL-1beta effects are mediated via CER generation, several follow-up studies were unable to detect any significant increase in CER levels in isolated beta cell following exposure to IL-1beta. For example, initial studies by Kwon et al. [24] reported virtually no effects of either IL-1beta or TNF-alpha on sphingomyelin hydrolysis in the beta cell. Further studies by these investigators demonstrated that while exogenously added cell-permeable analog of CER mimicked IL-1beta-mediated effects on beta cell dysfunction, CER, by itself, was unable to exert any demonstrable effects on iNOS expression and NO release. Along these lines Saldeen et al. [25] have also reported no significant effects of either IL-1beta or TNF-alpha on SMase activities in insulin-secreting RINm5F cells. In the same studies, these investigators observed that CER and TNF-alpha potentiated IL-1beta-mediated effects on iNOS expression, and that such potentiating effects of TNF-alpha were suppressed by SR 33557, an acid SMase inhibitor. Taken together, the above findings tend to suggest divergent mechanism(s) of action of CER and cytokines such as IL-1beta and TNF-alpha in promoting metabolic dysfunction of the beta cell. Such a postulation gained further strength from the studies of Major et al. [26] who reported that cell-permeable analogues of CERs decreased metabolic cell viability in a manner akin to cytokines; they also provided further evidence to suggest that cytokine-induced effects are not mediated via sphingomyelin hydrolysis and CER generation; studies compatible with original findings of Kwon et al. [24].
More recent data from our laboratory clearly provided evidence for CER-induced metabolic dysfunction of the islet beta cell [6]. We identified direct effects of exogenously added cell-permeable CER, but not its inactive analog, on mitochondrial dysfunction in insulin-secreting cells, including loss in mitochondrial membrane potential, cytochrome c release, caspase activation, and subsequent demise of the beta cell. In these studies, we presented first evidence to suggest that CER activates a mitochondrial protein phosphatase 2A-like activity in insulin-secreting cells [6, 27], leading to alterations in mitochondrial membrane properties, by promoting dephosphorylation of specific mitochondrial proteins (e.g., Bcl2). Interestingly, we noticed that effects of exogenous CER virtually mimicked the effects of IL-1beta on beta cell mitochondrial membrane properties. In these studies, we have also demonstrated that exogenously added CER exerted no direct effects on basal iNOS expression and NO release in INS cells. In fact, we observed a decrease in NO release in the presence of CER under basal conditions, thus implicating no direct effects of CER on iNOS expression and NO release. In support of those findings, our current findings suggest that inhibition of either SMase or CER synthase afford no protective effects against IL-1beta-induced iNOS expression, NO release and loss in metabolic cell viability in insulin-secreting beta cells. Taken together, our current findings further support our formulation that IL-1beta-mediated effects on iNOS expression and NO release are not mediated via the activation of either SMases or CER synthase. Additional studies are needed to identify specific signaling pathways and corresponding effector proteins that determine the metabolic fate of the beta cell under the duress of cytokines and/or CER.
Acknowledgements
This research was supported by grants (to AK) from the Medical Research Service of the Department of Veterans Affairs, the National Institute of Health (DK-56005) and the American Diabetes Association. AK is the recipient of Research Career Scientist Award from the Department of Veterans Affairs. GRJ is the recipient of Rumble Pre-Doctoral Fellowship from Wayne State University. RV and GRJ equally contributed to this work
References
- Corbett JA, Sweetland MA, Wang JL, Lancaster JR, McDaniel ML. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. ProcNatlAcadSci USA 1993; 90:1731-5 [PMID 8383325]
- Cunningham JM, Green IC. Cytokines, nitric oxide and insulin-secreting cells. Growth Regul 1994; 4:173-80. [PMID 7756973]
- Kowluru A, Morgan NG. GTP-binding proteins in cell survival and demise: the emerging picture in the pancreatic beta cell. BiochemPharmacol2002 ;63:1027-35. [PMID 11931834]
- Chen HQ, Tannous M, Veluthakal R, Amin R, Kowluru A. Novel roles for palmitoylation of Ras in IL-1beta-induced nitric oxide release and caspase activation in insulin-secreting beta cells. Biochem. Pharmacol 2003; 66:1681-94. [PMID 14563479]
- Veluthakal R, Chvyrkova I, Tannous M, McDonald P, Hadden T, Thurmond D, et al. Essential role for membrane lipid rafts in IL-1beta-induced nitric oxide release from insulin-secreting cells: potential regulation by caveolin-1. Diabetes 2005; 54:2576-85. [PMID 16123345]
- Veluthakal R, Palanivel R, Zhao Y, McDonald P, Gruber S, Kowluru A. Ceramide induces mitochondrial abnormalities in insulin-secreting INS-1 cells: potential mechanisms underlying ceramide-mediated metabolic dysfunction of the beta cell. Apoptosis 2005; 10:841-50. [PMID 16133874]
- Veluthakal R, Amin R, Kowluru A. Interleukin-1 beta induces posttranslational carboxymethylation and alterations in subnuclear distribution of lamin B in insulin-secreting RINm5F cells. Am J Physiol Cell Physiol 2004; 287:C1152-62. [PMID 15201138]
- Tannous M, Amin R, Popoff MR, Fiorentini C, Kowluru A. Positive modulation by Ras of interleukin-1beta-mediated nitric oxide generation in insulin-secreting clonal beta (HIT-T15) cells. BiochemPharmacol 2001; 62:1459-68. [PMID 11728382]
- Kwon G, Corbett JA, Rodi CP, Sullivan P, McDaniel ML. Interleukin 1beta-induced nitric oxide synthase expression by rat pancreatic beta cells: evidence for the involvement of nuclear factor kappa B in the signaling mechanism. Endocrinology 1995; 136:4790-5. [PMID 7588208]
- Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, et al. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol 2004; 164:123-31. [PMID 14709545]
- Won JS, Im YB, Khan M, Singh AK, Singh I. The role of neutral sphingomyelinase produced ceramide in lipopolysaccharide-mediated expression of inducible nitric oxide synthase. J Neurochem 2004; 88:583-93. [PMID 14720208]
- Lister MD, Ruan ZS, Bittman R. Interaction of sphingomyelinase with sphingomyelin analogs modified at the C-1 and C-3 positions of the sphingosine backbone. BiochimBiophysActa 1995; 1256:25-30. [PMID 7742352]
- Pilane CM, LaBelle EF. NO induced apoptosis of vascular smooth muscle cells accompanied by ceramide increase. J Cell Physiol 2004; 199:310-5. [PMID 15040013]
- Xu J, Yeh CH, Chen S, He L, Sensi SL, Canzoniero LMT, et al. Involvement of de novo ceramide biosynthesis in tumor necrosis factor-alpha/cycloheximide-induced cerebral endothelial cell death. J BiolChem 1998; 273:16521-6. [PMID 9632721]
- Lovat PE, Di Sano F, Corazzari M, Fazi B, Donnorso RP, Pearson AD, et al. Gangliosides link the acidic sphingomyelinase-mediated induction of ceramide to 12-lipoxygenase-dependent apoptosis of neuroblastoma in response to fenretinide. J Natl Cancer Inst 2004, 96:1288-99. [PMID 15339967]
- Desai K, Sullards MC, Allegood J, Wang E, Schmelz EM, Hartl M, et al. Fumonisins and fumonisin analogs as inhibitors of ceramide synthase and inducers of apoptosis. BiochimBiophysActa 2002; 1585:188-92. [PMID 12531553]
- Erdreich-Epstein A, Tran LB, Bowman NN, Wang H, Cabot MC, Durden DL, et al. Ceramide signaling in fenretinide-induced endothelial cell apoptosis. J BiolChem 2002; 277:49531-7. [PMID 12388538]
- Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis; a link between obesity and diabetes. ProcNatlAcadSci USA 1998; 95:2498-502. [PMID 9482914]
- Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J BiolChem 2003; 278:30015-21. [PMID 12771145]
- Kowluru A. Novel regulatory roles for protein phosphatase 2A in the islet beta cell. BiochemPharmacol 2005; 69:1681-91. [PMID 15935144]
- Sjoholm A. Ceramide inhibits pancreatic beta-cell insulin production and mitogenesis and mimics the actions of interleukin 1beta. FEBS Lett 1995; 367:283-6. [PMID 7607324]
- Sjoholm A. Aspects of the involvement of interleukin-1 and nitric oxide in the pathogenesis of insulin-dependent diabetes mellitus. Cell Death Differ 1998; 5:461-8. [PMID 10200497]
- Welsh N. Interleukin-1 beta-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 in the insulin-producing cell line RINm5F. J BiolChem 1996; 271:8307-12. [PMID 8626526]
- Kwon G, Bohrer A, Han X, Corbett JA, Ma Z, Gross RW, et al. Characterization of the sphingomyelin content of isolated pancreatic islets. Evaluation of the role of sphingomyelin hydrolysis in the action of interleukin-1 to induce islet overproduction of nitric oxide. BiochimBiophysActa 1996; 1300:63-72. [PMID 8608164]
- Major CD, Gao ZY, Wolf BA. Activation of the sphingomyelinase/ceramide signal transduction pathway in insulin-secreting beta-cells: role in cytokine-induced beta cell death. Diabetes 1999; 48:1372-80. [PMID 10389841]
- Saldeen J, Jaffrezou JP, Welsh N. The acid sphingomyelinase inhibitor SR33557 counteracts TNF-alpha-mediated potentiation of IL-1beta-induced NF-kappaB activation in the insulin-producing cell line Rinm5F. Autoimmunity 2000; 32:241-54. [PMID 11191283]
- Jangati GR, Veluthakal R, Kowluru A. siRNA-mediated depletion of endogenous protein phosphatase 2Acalpha markedly attenuates ceramide-activated protein phosphatase activity in insulin-secreting INS-832/13 cells. BiochemBiophys Res Commun 2006; 348:649-52. [PMID 16884689]

