Kate M Ellery and Steven H Erdman
Division of Gastroenterology, Hepatology, and Nutrition, Nationwide Children’s Hospital, The Ohio State University College of Medicine, Columbus, Ohio, USA
- *Corresponding Author:
- Kate M. Ellery, DO
700 Children’s Drive JW 1988
Columbus
Ohio 43205
USA
Phone: 614.722.3450
Fax: 614.722.3454
Email kate.ellery@nationwidechildrens.org
Received April 15th, 2014 – Accepted May 29th, 2014
Keywords
Ectodermal Dysplasia; Exocrine Pancreatic Insufficiency; Johanson Blizzard syndrome
INTRODUCTION
Johanson-Blizzard Syndrome (JBS) (OMIM #243800) is a rare autosomal recessive disorder first described in 1971 by Johanson and Blizzard [1]. Recent review of the medical literature identifies fewer than 100 cases reported worldwide, primarily in consanguineous unions [1, 2]. The syndrome is most classically characterized by severe congenital exocrine pancreatic insufficiency and hypoplasia or aplasia of the nasal alae. Additional features include: short stature (>80%), dental abnormalities (>80%), sensorineural hearing loss (80%), mental retardation (77%), scalp defects including alopecia (76%), hypothyroidism (40%), imperforate anus (39%), and genitourinary malformations (38%) [2]. Cardiac defects have been reported [3-5]. Neonatal or early exocrine pancreatic insufficiency is thought to be a consistent finding in JBS. Abdominal imaging of affected patients shows replacement of the exocrine pancreas by lipomatous and connective tissues [1, 2]. Autopsy studies reveal a significant deficit in pancreatic acinar cells; however, the islets of Langerhans remain intact [6]. This defect leads to almost complete absence of pancreatic enzymes in duodenal secretions resulting in maldigestion and poor absorption. Here we present a 15 year old male with the onset of pancreatic insufficiency in mid-adolescence with report of a similarly affected identical twin.
CASE REPORT
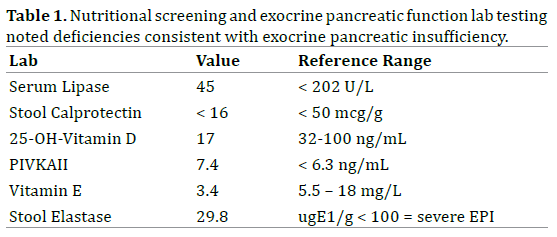
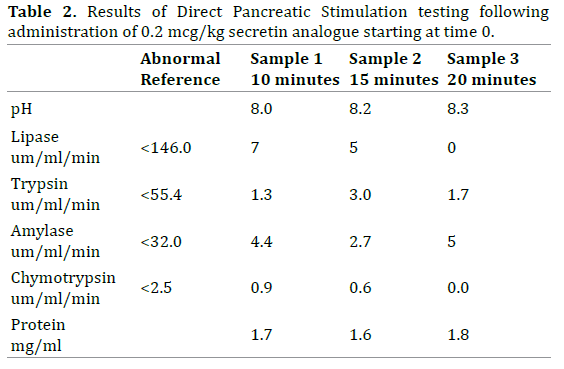
The patient, a well-nourished adolescent with prior diagnosis of ectodermal dysplasia, presented at 15 years of age with sudden onset of oily diarrhea (10-15 stools/ day) and rapid weight loss. Examination revealed alopecia, thick yellow nails (Figure 1), small malformed teeth and subtle nasal alae hypoplasia. Initial evaluation for gastrointestinal infection, celiac disease and inflammatory bowel disease was unremarkable including upper and lower endoscopy. Nutritional screening revealed fatsoluble vitamin deficiencies and a fecal elastase level indicating severe exocrine pancreatic insufficiency (Table 1). An abdominal ultrasound showed a uniform, echogenic and atrophic pancreas with fatty replacement. Direct pancreatic stimulation testing using a method similar to that described by Wali et al. revealed nearly absent pancreatic enzyme secretion (Table 2) [7]. Subsequent pancreatic enzyme replacement therapy (Creon™ 24K capsules; 3,000 lipase units/kg/day) and fat-soluble vitamin supplementation (ADEKS™ tablets, 1 BID) resulted in significant weight gain (Figure 2) and resolution of oily stools. The patient and family have repeatedly declined genetic testing.
Figure 1. Thick yellow toe nails consistent with ectodermal dysplasia
from our patient.
Figure 2. Weight for Age plot focusing on age 10 years to last weight
measurement. The arrow denotes the start of pancreatic enzyme
replacement and fat-soluble vitamin supplementation.
The patient has an identical twin brother with ectodermal dysplasia, a thin body habitus and reported chronic diarrhea beginning at 16 years of age. The brother declined all formal medical evaluation.
DISCUSSION
Classic cases of JBS present in early infancy with syndromic features and severe exocrine pancreatic insufficiency. The genetic defect of JBS is a homozygous loss-of-function mutation in the Ubiquitin-Protein Ligase E3 Component N-Recognin 1 (UBR1) gene [1, 5]. This gene is highly expressed in pancreatic tissue [3] where it functions primarily in the regulation of protein degradation, control of the levels of many intracellular proteins and perhaps as a defense against noxious stimuli [2]. UBR1 protein deficiency contributes to the gradual destruction of previously formed acinar cells resembling pancreatitis of intrauterine onset [3]. Nonsense, frame shift and splicesite mutations have been described in classic, severe cases [1]; whereas, missense or in-frame deletions are thought to lead to less severe phenotypic presentations of JBS [3, 8, 9].
Previous case reports have described heterozygous deletions or missense mutations with resultant partial or residual UBR1 activity and pancreatic function; this has been thought to explain the reported mild phenotypes of this disease [6, 8]. Variable expressivity of the same mutation within a family is well documented [3].
Our patient presented with sudden onset of pancreatic insufficiency as demonstrated by acute weight loss, oily stools and fat-soluble vitamin malabsorption at 15 years of age. We hypothesize that an existing subtle phenotype in conjunction with a progressive destructive process of the pancreas ultimately resulted in exocrine pancreatic insufficiency. Infectious, environmental or epigenetic factors may influence the timing of these phenotypic events. It is thought that an ongoing destructive process plays a role in the development of endocrine insufficiency and insulin deficiency later in life [1-3, 6].
CONCLUSION
Johanson-Blizzard Syndrome is a rare autosomal recessive form of congenital pancreatic insufficiency. This case illustrates that exocrine pancreatic insufficiency can occur as an evolving process manifesting later in life. Missense, in-frame deletions and heterozygous mutations which have differing, less pathologic effects likely explain the milder phenotypes of this disease [3, 8, 9]. It is important to recognize the phenotypic variations of this syndrome when caring for those with ectodermal dysplasia who may initially lack the typical findings of exocrine pancreatic insufficiency, as there may be more patients with milder phenotypes than previously identified.
Acknowledgements
We would like to thank Drs. Cheryl Gariepy, Jennifer Dotson and Soma Kumar for their thoughtful review and comments on this case report.
Conflicts of Interest
The authors have no conflicts or disclosures.
References
- Rezaei N, Sabbaghian M, Liu Z, Zenker M. Eponym: Johanson-Blizzard syndrome. Eur J Pediatr. 2011; 170: 179-83. [PMID: 20556422]
- Zenker M, Mayerle J, Reis A, Lerch MM. Genetic basis and pancreatic biology of Johanson-Blizzard syndrome. EndocrinolMetabClin North Am. 2006; 35: 243-53, vii-viii. [PMID: 16632090]
- Schoner K, Fritz B, Huelskamp G, Louwen F, Zenker M, et al. Recurrent Johanson-Blizzard syndrome in a triplet pregnancy complicated by urethral obstruction sequence: a clinical, molecular, and immunohistochemical approach. PediatrDevPathol. 2012; 15: 50-7. [PMID: 21711208]
- Hurst JA, Baraitser M. Johanson-Blizzard syndrome. J Med Genet. 1989; 26: 45-8. [PMID: 2645405]
- Almashraki N, Abdulnabee MZ, Sukalo M, Alrajoudi A, Sharafadeen I, et al. Johanson-Blizzard syndrome. World J Gastroenterol. 2011; 17: 4247- 50. [PMID: 22072859]
- Alkhouri N, Kaplan B, Kay M, Shealy A, Crowe C, et al. Johanson- Blizzard syndrome with mild phenotypic features confirmed by UBR1 gene testing. World J Gastroenterol. 2008; 14: 6863-6. [PMID: 19058315]
- Wali PD, Loveridge-Lenza B, He Z, Horvath K. Comparison of fecal elastase-1 and pancreatic function testing in children. J PediatrGastroenterolNutr. 2012; 54: 277-80 [PMID: 22266489]
- Hwang CS, Sukalo M, Batygin O, Addor MC, Brunner H, et al. Ubiquitin ligases of the N-end rule pathway: assessment of mutations in UBR1 that cause the Johanson-Blizzard syndrome. PLoS One. 2011; 6: e24925. [PMID: 21931868]
- Elting M, Kariminejad A, de Sonnaville ML, Ottenkamp J, Bauhuber S, et al. Johanson-Blizzard syndrome caused by identical UBR1 mutations in two unrelated girls, one with a cardiomyopathy. Am J Med Genet A. 2008; 146A: 3058-61. [PMID:19006206]



