Keywords
Polysiloxane; Thermogravimetric analysis; Degradation; Inert and oxygen atmosphere; Kinetics
Introduction
One of the technically most important advantages of polysiloxanes is their superior thermal stability if compared to the other polymeric materials. Their applicability exhibits over the wide temperature range. Beside the high thermal stability, polysiloxanes also possess low electrical conductivity and a large degree of main-chain flexibility. For that reason their value is recognized in different areas, namely automotive industry, metallurgy, electronics and medicine, used as in a form of lubricants, high temperature stable fluids, elastomers or adhesives. The most commercially important polysiloxane, and also the simplest one (having a Si–O backbone and two methyl substituent per silicon atom), is polydimethylsiloxane (PDMS) [1,2].
One of the most frequently used methods for preparing polysiloxane emulsions, which are often employed as lubricants in die casting process of aluminum alloys, is ring-opening polymerization (ROP) of cyclosiloxanes in emulsion. Comparing the ROP polymerization in emulsion to bulk or solution ROP, the ROP in emulsion gives higher monomer conversion and products of higher molecular weight with narrower distribution [3-5]. Moreover, by emulsion ROP method polymer of designed molecular weight distribution can be obtained. However, to obtain stable emulsions of desired rheological properties a number of different anionic, cationic and nonionic emulsifiers have been used in different ROP polymerization recipes for polysiloxane emulsions production. The main drawback of obtained emulsions is their emulsifier and initiator residues content, which can affect polysiloxane thermal stability [4,6-11].
For anionic ROP (AROP) of cyclosiloxanes in emulsion cationic and non-ionic emulsifiers are typically employed. The cationic emulsifier is actively involved in the reaction mechanism of AROP in emulsion, which is known for its complex mechanism, since different chemical reactions take place on the polymer particle surface and inside the particle. However, main chemical reactions of polymer formation (initiation, propagation and termination) occur on the particle surface. When a growing polymer chain reaches a critical degree of polymerization and its termination takes place, the chain penetrates into the particle where condensation and redistribution reactions occur. The extents of condensation and redistribution reactions affect the average molecular weight and distribution of the polymer drastically. In order to achieve a narrow molecular weight distribution, much attention to control of backbiting, condensation and redistribution reactions has to be payed [3,5,12]. On the other hand, condensation reactions are desirable when polymer of higher average molecular weight is preferred. The condensation reactions were proven by a study of De Gunzbourg et al. [3,13], who observed a significant increase in average molecular weight and polydispersity index above 70 and 60% of monomer conversion.
Thermal properties and thermal degradation of polysiloxanes have been studied extensively over the past decades [14-22]. It was proven that the progress of degradation and obtained degradation products depend on atmosphere, since different mechanisms of degradation in different atmospheres occur, and on the presence of impurities, which can catalyze/initiate the degradation process. Degradation in an inert atmosphere, such as N2, results in depolymerization in which volatile cyclic oligomers of low molecular weight are formed. In inert atmosphere polysiloxanes generally degrade by a single weight-loss step (TGA) and without coloration of the residue. Depolymerization under high temperature may be explained by three mechanisms proposed in the literature, known as unzipping mechanism [15,17], random scission mechanism [15,17] and external catalyzed mechanism [15] (presence of ionic and polar impurities or additives). On the other hand, thermal degradation of polysiloxanes in the presence of oxygen follows predominantly the radical mechanism. Along with the cyclic oligomers, also other volatile products during polymer decomposition are formed. For that reason a typical TGA experiment shows a two-step process, where one step in general starts at the same temperature as in the case of inert atmosphere, and the other is associated with volatile products typically consisted of carbon monoxide/ oxide, water, formaldehyde, methanol and traces of formic acid. The residue of such experiment is silica, which is a result of polysiloxanes oxidation through natural tendency of silicon to bond with oxygen [14,15,17].
The main object of the proposed study is to prepare PDMS samples with different molecular weights and to study the effect of molecular weight on the thermal stability of PDMS in both, inert and oxidative atmosphere. Realistic PDMS samples containing emulsifiers and initiator residues, similar to those used as lubricants in die casting process of aluminum alloys, of different molecular weights were prepared by AROP in emulsion. Their thermal stability was investigated using TGA.
Materials and Methods
Materials
Octamethylcyclotetrasiloxane (D4, Aldrich, 98%) was used as received without further purification. Hexadecyltrimethylammonium bromide (CTAB, Aldrich, ≥98%) and secondary alcohol ethoxylate (Tergitol, Type 15-S-9, p.a., Aldrich) were used as cationic and nonionic surfactants, respectively, in the recipe for D4 emulsion polymerization, where potassium hydroxide (p.a., Merck) was used as initiator. Hydrochloric acid (p.a., Merck, 0.5 M) was used in order to stop the polymerization reaction. Water was previously deionized for use.
Sample Preparation
Polysiloxane emulsions were synthesized isothermally at 80°C by batch AROP in emulsion using a 250 ml glass reactor with four necks equipped with a reflux condenser, a mechanical stirrer, a digital thermometer, and a nitrogen gas inlet [23]. Monomer pre-emulsion was prepared at room temperature by dissolution of ionic (5.63 g) and nonionic emulsifier (0.63 g) in deionized water (300 g) in the reactor. Monomer (50 g) was added to the emulsifier solution and the pre-emulsion was stirred at 1000 rpm for 15 minutes. Then the mixture was heated up to 80°C and the stirring speed lowered to 500 rpm. To initiate the polymerization KOH (1.0 g) was added. To obtain PDMS samples of different molecular weights the reactor content was sampled during polymerization. To stop the polymerization in the withdrawn samples the pH value was adjusted to 7.5-8 using HCl (0.5 M).
D4 conversion of withdrawn samples was determined gravimetrically. The samples were dried to constant mass at 50°C in vacuum oven. During vacuum exposure all the volatile components, monomer and small oligomeric cycles, were removed from the sample. Thus, only the amount of monomer converted to large, non-volatile polymeric chains was determined and the monomer conversion should be interpreted accordingly. Since the study was based on a realistic system, where high purification costs were not tolerable, prepared samples were not further purified via chromatography. For that reason the presence of emulsifiers was considered in conversion calculations of samples taken during polymerization.
Average molecular weights of polymer in withdrawn samples were determined by size exclusion chromatography (SEC) using polystyrene standards (between 1,060 and 750,000 g mol–1). Hewlett Packard 1100 Series instrument equipped with MIXED-D column (5 μm, Polymer Laboratories) coupled with refractiveindex detector (Agilent Technologies) were applied. The 0.2% (w/V) solutions were prepared in toluene, which was also used as carrier solvent at a rate of 1 ml min–1. Before SEC the unreacted monomer and emulsion water were removed from samples using a vacuum dryer at 50°C.
Thermogravimetric analysis
Thermal stability of PDMS samples (after unreacted monomer and emulsion water removal) was assessed by thermogravimetric analysis (TGA). Before TGA the unreacted monomer and emulsion water were removed from samples using a vacuum dryer at 50°C. TGA measurements were performed in nitrogen and oxygen atmospheres with a gas flow rate of 50 ml min–1 using a Mettler Toledo (Schwerzenbach, Switzerland) TGA/DSC1 instrument. The investigated temperature range was from 25 up to 700°C. Heating rate of 10 K min–1 was used. For measurements standard 70 μL Al2O3 crucibles were used and the baseline was automatically subtracted. The sample mass was around 10 mg.
After TGA analysis, residuals were investigated by Fourier transform-infrared spectroscopy (FTIR) using KBr disks on Perkin Elmer Spectrum 1000 spectrometer.
Results and Discussion
Preparation of PDMS samples of different molecular weights
AROP of D4 in emulsion involves three reactions, which affect the monomer consumption: initiation, propagation and reversible termination. As it was stated by De Gunzbourg [3] and Zhang [7] and confirmed by others [12,24,25] all three reactions occur at the particle/water interface, where also backbiting reactions take place. The latter are responsible for low final conversions [12,24,25]. Using the AROP procedure described in the experimental part a final monomer conversion of 84.5% (Figure 1) was obtained. In Figure 1, in which monomer conversion evolutions with time for two replicates of PDMS synthesis are shown, the process reproducibility was confirmed.
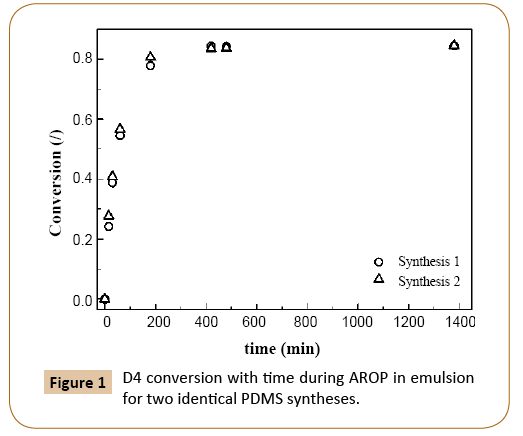
Figure 1: D4 conversion with time during AROP in emulsion for two identical PDMS syntheses.
Molecular weight of PDMS increased with monomer conversion and continued to increase after equilibrium monomer conversion of 84.5% was reached (Figure 2 and Table 1). In reversible termination reaction, the active oxygen of the growing PDMS chains reacts with water to give silanol end-group. As mentioned before, when such terminated polymer chain reaches a critical degree of polymerization, the chain penetrates into the particle interior [3,5], where condensation and redistribution reactions take place, which affect the polymer molecular weight and distribution. The condensation reactions are responsible for further increase of the PDMS molecular weight at unchanged conversion. This may be confirmed by a comparison of shapes of molecular weight distribution curves in Figure 2, where peak onsets shift toward lower elution volumes with reaction time while the offsets remain nearly similar. The average molecular weights of obtained PDMS samples are collected in Table 1.
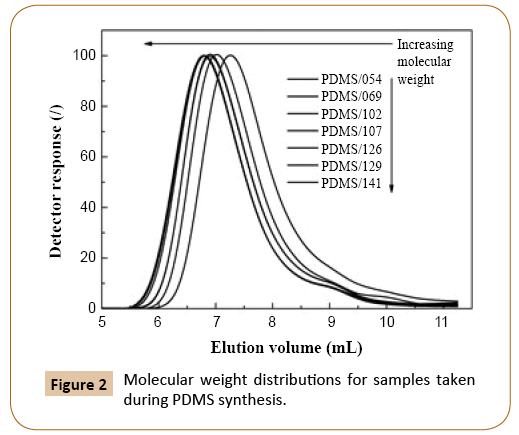
Figure 2: Molecular weight distributions for samples taken during PDMS synthesis.
Table 1 D4 conversion and PDMS molecular weights of selected samples.
| Time (min) |
D4 conversion (%) |
Mn × 103 (/) |
Mw × 103 (/) |
Sample name |
| 60 |
55 |
5.4 |
30.9 |
PDMS/054 |
| 180 |
79 |
6.9 |
46.6 |
PDMS/069 |
| 420 |
84 |
10.2 |
57 |
PDMS/102 |
| 480 |
84 |
10.7 |
57.5 |
PDMS/107 |
| 1380 |
84 |
12.6 |
72.5 |
PDMS/126 |
| 1560 |
85 |
12.9 |
75.8 |
PDMS/129 |
| 1740 |
85 |
14.1 |
79.2 |
PDMS/141 |
Mn – number average molecular weight. MW – weight average molecular weight.
Thermal stability of PDMS samples of different molecular weights PDMS samples of known average molecular weight, which are collected and labeled in Table 1, were analyzed by TGA as described in the experimental section. After drying, samples contained PDMS and emulsifiers and initiator residuals. The amount of emulsifiers and initiator in a dried sample depended on the monomer conversion; at higher monomer conversion, the amount of impurities was lower. The thermal stability of emulsifiers was lower than that of PDMS, therefore different mass loss steps in TGA thermograms could be sensibly attributed to specific compounds, i.e. to emulsifiers and PDMS (Figure 3). It may be estimated that emulsifiers decomposed completely at temperature below 450°C, while PDMS decomposed at around 550°C.
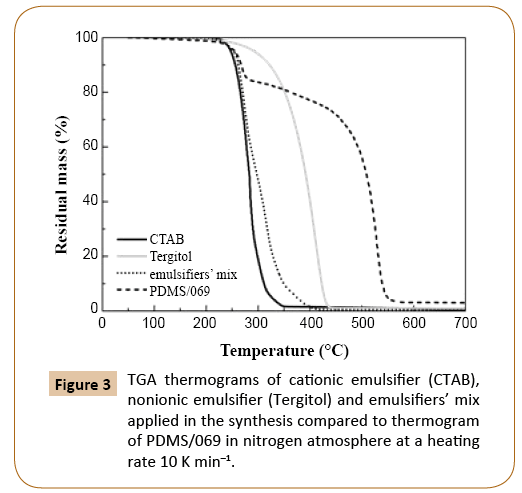
Figure 3: TGA thermograms of cationic emulsifier (CTAB), nonionic emulsifier (Tergitol) and emulsifiers’ mix applied in the synthesis compared to thermogram of PDMS/069 in nitrogen atmosphere at a heating rate 10 K min–1.
In Figures 4 and 5 TGA thermograms for PDMS samples of different molecular weights in nitrogen and oxygen atmospheres are shown, respectively. The weight loss signal below 400°C was attributed to emulsifiers’ mix degradation. The weight loss due to degradation of emulsifiers decreased with increasing molecular weight of PDMS since the amount of emulsifiers in samples decreased with increasing monomer conversion. As it has been already reported in the literature [16,26], the degradation in oxygen atmosphere started at lower temperature than in the nitrogen atmosphere (Figures 4 and 5), when sample was exposed to the same heating rate.
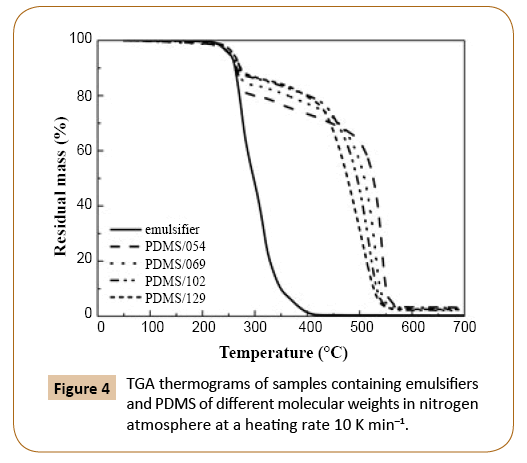
Figure 4: TGA thermograms of samples containing emulsifiers and PDMS of different molecular weights in nitrogen atmosphere at a heating rate 10 K min–1.

Figure 5: TGA thermograms of samples containing emulsifiers and PDMS of different molecular weights in oxygen atmosphere at a heating rate 10 K min–1.
In nitrogen atmosphere (Figure 4), the molecular weight of PDMS affected the shape of TGA curve drastically at higher temperature. The lower the molecular weight the better the thermal stability of PDMS. For PDMS of lower molecular weights the predominant decomposition mechanism is the unzipping mechanism, since the concentration of reaction sites involved in this mechanism, i.e. -Si-OH ends, is higher than in the case of PDMS of higher molecular weights. For that reason the degradation of PDMS also starts at lower temperatures in the case of lower molecular weights. However, when the molecular weight increased, the temperature interval of degradation became narrower, which could be associated with the lower thermal stability of PDMS. The observed behavior could be attributed to the increased probability of random scission of polymer chains of higher molecular weight, caused by intramolecular and intermolecular redistribution reactions. Depolymerization as a result of redistribution reactions is faster than depolymerization by unzipping mechanism, which could also explain the reason of better thermal stability of PDMS samples with lower molecular weights. However, it has to be noticed that also external catalyzed mechanism could affect the degradation of PDMS, since impurities in the sample cause the bond cleavage of Si-O with the binding of OH on Si atom. With the higher amount of impurities in the samples higher effects of external catalyzed mechanism would be expected. Conversely, sample PDMS/129 with the lowest thermal stability had the lowest amount of impurities, whereas PDMS/054 with the highest thermal stability contained the highest amount of impurities (Figure 4). Consequently, there is no clear trend indicating the effect of impurities on the thermal stability of samples. The observed is in disagreement when considering that different amount of impurities would significantly affect the degradation behavior. Therefore, it can be concluded that external catalyzed mechanism was not predominant degradation mechanism and had negligible effect on PDMS thermal stability and degradation behavior. After the PDMS degradation step, the residual mass was minimal, indicating that decomposition products were volatile. The residue was composed mainly of mineral salts, due to neutralization with HCl after emulsion sampling during synthesis. The amount of residue was similar for all samples.
In oxygen atmosphere, the PDMS samples degraded in the same temperature interval regardless of their molecular weight (Figure 5). The only difference between the thermograms is the consequence of different amounts of emulgators and inorganic salts in the sample and not the difference in the molecular weight. After PDMS degradation step the amount of non-volatile residue was high. The residue consisted mainly of SiO2, which is formed by free radical decomposition mechanism in oxygen atmosphere.
Kinetic model of thermal degradation in nitrogen atmosphere
Due to the fact that the thermal degradation in inert atmosphere is a function of molecular weight, we tried to describe such behavior in a form of a kinetic model in order to predict the thermal degradation of polydimethylsiloxane related to its molecular weight without performing thermogravimetric analysis. When building kinetic model we focused on two main effects: degradation of polydimethylsiloxane and evaporation of the formed molecules. For that reason apparent activation energy for the first and the second process was obtained according to the literature [16]. Camino et al. [16] suggested that at the heating rates of 10 K min–1 or higher diffusion-limited kinetics prevailed. Accordingly, thermal degradation of PDMS in diffusion-limited kinetics can be eventually described as evaporation rates of the degradation products. Consequently, in our calculations we used the evaporation energy of the smallest (and presented in the highest amount) among degradation products - hexamethylcyclotrisiloxane, which is known to be 55.3 kJ mol–1. The value was considered to be the lowest limit of the Eapp1 term in eqn. (1), representing a temperature dependence of diffusion/evaporation process [16]. Furthermore, apparent activation energy covering depolymerization of PDMS and formation of intramolecular, cyclic transition state as the rate-determining step was predicted to be 167.9 kJ mol–1 [16]. Nevertheless, first the modification of experimental data for further usage in model fitting was performed. Since the part of residual mass drop as a result of emulsifiers’ evaporation at around 300°C was not directly linked to the thermal degradation of PDMS (Figure 4), the appropriate amount of residual mass was subtracted from the rest of experimental data. Thereafter the rest of the experimental data werenormalized within (0, 1) range. Finally, in least squares approximation between the experimental data and calculated data, the model equation eqn. (1) was proposed first.
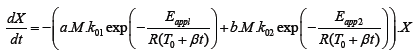 (1)
(1)
In eqn. (1) X (relative degree of degradation) is the normalized value and represents the proportion of Si-O related to the overall amount of Si-O at the beginning of the reaction. When the thermal degradation proceeds, the portion of remaining Si-O in the sample starts to decrease as a consequence of thermal degradation and evaporation. The proceeding of thermal degradation is regulated by molecular mass M, since the thermal degradation in inert atmosphere is dependent on its molecular weight as already demonstrated. The order of the magnitude and the correctness of the units is adjusted by factors a and b in such a way that their product with molecular weight remained without units. The reaction rate is described by Arrhenius equation, where k01 and k02 are pre-exponential factors for diffusion-limited kinetics and for cyclization of the degradation products in s-1, respectively. Furthermore Eapp1 and Eapp2 are apparent activation energies for diffusion/evaporation controlled process and rate-limiting kinetics, respectively, both in J mol–1, R is the ideal gas constant in J mol–1 K–1, t is time in seconds, T0 is initial temperature (ambient temperature) in K and β is the heating rate in K min–1.
Despite the fact that the results of the fitting showed poor agreement between experimental and calculated data with R2 of 0.9730, especially in the region with the fast drop of the remaining Si-O, we made further adjustment to the molecular weight integer by factor n in diffusion-limited kinetics section of the equation and factor m in depolymerization section of the equation (eqn. (2)). As in the previous case, the products of a. Mn and b. Mm were considered to be without units.
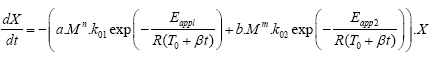 (2)
(2)
The results of model fitting using eqn. (2) and achieving R2 0.9916 are presented in Figure 6. It can be seen that satisfying agreement between experimental and calculated data was achieved with a slight deviation for the sample PDMS/054, presumably due to its broader molecular weight distribution comparable to other samples as seen in Figure 2. Among that, the peak of the sample with the lowest molecular mass appeared at elution volume, which in not really comparable to the trend of other samples. Although we previously discussed and distinguished between the unzipping and intramolecular/intermolecular redistribution as the reason for differences in polymerization in inert atmosphere, mentioned in eqn. (2) is not unambiguously represented. Nevertheless, the effect of unzipping degradation mechanism accompanied with intramolecular/intermolecular redistribution is considered in the product of a. Mn and b. Mm.
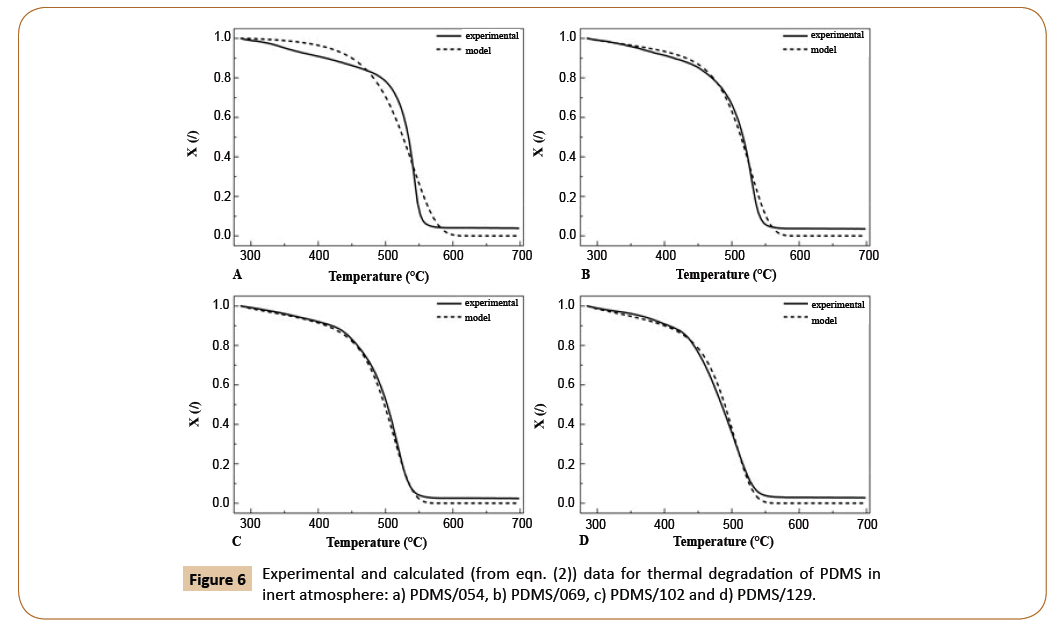
Figure 6: Experimental and calculated (from eqn. (2)) data for thermal degradation of PDMS in inert atmosphere: a) PDMS/054, b) PDMS/069, c) PDMS/102 and d) PDMS/129.
The result of the mathematical modeling is the final model equation of thermal degradation of polydimethylsiloxane considering the variation of molecular weight (eqn. (3)).
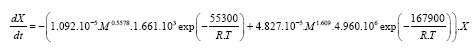 (3)
(3)
The obtained equation is relevant for molecular weights from 54000–129000 at a heating rate of 10 K min–1 in order to predict the degradation of PDMS in nitrogen atmosphere.
Conclusions
The effects of polydimethylsiloxane molecular weight on its thermal stability were investigated in inert and oxygen atmosphere. The results in inert atmosphere showed better thermal stability of samples with lower molecular weights, which we explained by different mechanisms of thermal degradation of PDMS. The predominant mechanism at lower molecular weights is unzipping mechanism, since the amount of -Si- OH end molecules and consequently the amount of reaction sites involved in unzipping mechanism is higher than in the case of higher molecular weights. At higher molecular weights redistribution reactions are predominant and are faster than depolymerization under unzipping mechanism, which affect the thermal stability of PDMS. Experiments in oxygen atmosphere showed no effect of polydimethylsiloxane molecular weight on its thermal stability. Eventually we proposed a kinetic model, which describes the behavior of thermal degradation in inert atmosphere considering its molecular weight. Proposed kinetic model showed satisfying agreement between experimental and calculated data and appropriate kinetic parameters were derived as a result.
Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (research core funding No. P2-0191). The authors acknowledge the project (Multifunctional Bio-based Hybrid Thermosets: Synthesis, Characterization and Potential Use in Composites, ID N2-0033) was financially supported by the Slovenian Research Agency.
References
- Lewis RN (1948) Methylphenylpolysiloxanes. Journal of the American Chemical Society 70: 1115-1117.
- Dvornic PR (2000) Thermal properties of polysiloxanes. In Silicon-containing polymers. Springer Netherlands pp: 185-212.
- De Gunzbourg A, Favier JC, Hémery P (1994) Anionic polymerization of octamethylcyclotetrasiloxane in aqueous emulsion I: Preliminary results and kinetic study. Polymer international 35: 179-188.
- Barrère M, Maitre C, Dourges MA, Hémery P (2001) Anionic polymerization of 1,3,5-tris (trifluoropropylmethyl) cyclotrisiloxane (F3) in miniemulsion. Macromolecules 34: 7276-7280.
- Barrere M, Ganachaud F, Bendejacq D, Dourges MA, Maitre C, et al. (2001) Anionic polymerization of octamethylcyclotetrasiloxane in miniemulsion II. Molar mass analyses and mechanism scheme. Polymer 42: 7239-7246.
- Landfester K, Bechthold N, Tiarks F, Antonietti M (1999) Miniemulsion polymerization with cationic and nonionic surfactants: a very efficient use of surfactants for heterophase polymerization. Macromolecules 32: 2679-2683.
- Zhang D, Jiang X, Yang C (2003) Microemulsion polymerization of siloxane with nonionic surfactants as emulsifiers. Journal of applied polymer science 89: 3587-3593.
- Silas JA, Kaler EW, Hill RM (2001) Effect of didodecyldimethylammonium bromide on the phase behavior of nonionic surfactant− silicone oil microemulsions. Langmuir 17: 4534-4539.
- Maisonnier S, Favier JC, Masure M, Hémery P (1999) Cationic polymerization of 1,3,5,7â€ÂÂÂÂtetramethylcyclotetrasiloxane in aqueous emulsion. Part I. Preliminary results. Polymer international 48: 159-164.
- Palaprat G, Ganachaud F, Mauzac M, Hémery P (2005) Cationic polymerization of 2,4,6,8-tetramethylcyclotetrasiloxane processed by tuning the pH of the miniemulsion. Polymer 46: 11213-11218.
- Bischoff R, Sigwalt P (1996) Mechanism of octamethylcyclotetrasiloxane polymerization in the presence of siloxanediols. Polymer international 40: 99-109.
- MohoriÄ I, Šebenik U (2011) Anionic ring-opening polymerization of octamethylcyclotetrasiloxane in emulsion above critical micelle concentration. Polymer 52: 1234-1240.
- De Gunzbourg A, Maisonnier S, Favier JC, Maitre C, Masure M, et al. (1998) Ionic polymerization in aqueous emulsion. In Macromolecular Symposia, WILEYâ€ÂÂÂÂVCH Verlag GmbH & Co. KGaA 132: 359-370.
- Lewis CW (1958) The pyrolysis of dimethylpolysiloxanes. Journal of Polymer Science Part A: Polymer Chemistry 33: 153-159.
- Grassie N, Macfarlane IG (1978) The thermal degradation of polysiloxanes—I. Poly (dimethylsiloxane). European polymer journal 14: 875-884.
- Camino G, Lomakin SM, Lazzari M (2001) Polydimethylsiloxane thermal degradation Part 1. Kinetic aspects. Polymer 42: 2395-2402.
- Camino G, Lomakin SM, Lageard M (2002) Thermal polydimethylsiloxane degradation. Part 2. The degradation mechanisms. Polymer 43: 2011-2015.
- Jovanovic JD, Govedarica MN, Dvornic PR, Popovic IG (1998) The thermogravimetric analysis of some polysiloxanes. Polymer Degradation and Stability 61: 87-93.
- Liao SK, Jang SC, Lin MF (2007) Thermal degradation of imide-containing siloxane-urethane copolymer. Journal of polymer engineering 27: 197-218.
- Hanu LG, Simon GP, Cheng YB (2006) Thermal stability and flammability of silicone polymer composites. Polymer Degradation and Stability 91: 1373-1379.
- Tiwari A, Nema AK, Das CK, Nema SK (2004) Thermal analysis of polysiloxanes, aromatic polyimide and their blends. ThermochimicaActa 417: 133-142.
- Zhixiong H, Wenfeng X, Dongcai C (2006) The thermal decomposition kinetics of polysiloxane/polymethylacrylate IPNs materials. Journal of Wuhan University of Technology-Mater. Sci. Ed. 21: 47-49.
- MohoriÄ I, Krajnc M, Šebenik U (2009) Model-free kinetics analysis of thermal degradation of polysiloxane lubricant. Chemical and biochemical engineering quarterly 23: 493-496.
- MohoriÄ I, Šebenik U (2011) Semibatch anionic ring-opening polymerization of octamethylcyclotetrasiloxane in emulsion. Polymer 52: 4423-4428.
- MohoriÄ I, Šebenik U (2013) Semibatch anionic ringâ€ÂÂÂÂopening polymerization of octamethylcyclotetrasiloxane in emulsions: effect of the amount of seed polymer particles. Polymer International 62: 1022-1028.
- Hamdani S, Longuet C, Perrin D, Lopez-cuesta JM, Ganachaud F (2009) Flame retardancy of silicone-based materials. Polymer Degradation and Stability 94: 465-495.
