Keywords
Biological Markers; CA-19-9 Antigen; Pancreatic Neoplasms; Proteomics
Abbreviations
MALDI: matrix assisted laser desorption/ ionisation; MS mass spectrometry; SELDI: surface enhanced laser desorption ionization; TOF time of flight; UV: ultraviolet
Pancreatic cancer is the fourth leading cause of cancer death in the United States with approximately 34,000 deaths in 2008. Despite advances in understanding the pathogenesis of the disease, the five-year survival remains at 4%, with a majority of patients not surviving longer than one year [1]. As surgical resection remains the only reliable curative method, improving the 5-year survival to approximately 25%, early detection is paramount [2, 3, 4]. Delays in diagnosis are often due to small cancers or the presence of non specific symptoms. Whilst there have been significant developments in the imaging modalities for pancreatic lesions in the last two decades, they have not influenced the overall survival from pancreatic cancer [5].
Research over the last few years has identified certain genetic alterations associated with pancreatic cancer, such as point mutations of K-ras occurring in 90% of cases, and inactivation of other tumour suppressor genes such as p53 and p16 [6, 7]. Unfortunately to date these have not led to the development of new biomarkers. The current clinical tumour markers for pancreatic cancer, CEA and CA 19-9, lack the appropriate sensitivity and specificity required for screening an asymptomatic population to aid early diagnosis. CEA, a membrane glycoprotein, is less sensitive in the diagnosis of pancreatic cancer with sensitivities reported between 59-71% and specificities of 64-66%. In comparison, studies have reported a sensitivity ranging 79-89% and specificity of 72-90% for CA 19-9 [8]. Unfortunately, CA 19-9 has shown limited success in identifying small cancers with the Japanese cancer registry reporting only 48.4% of patients with pancreatic cancers less than 2 cm showing increased levels [9]. CA 19-9 levels are also elevated in both other malignancies and benign conditions such as pancreatitis and biliary obstruction [10]. Furthermore, 10% of the population possess a negative genotype for the Lewis gene and are therefore unable to produce CA 19-9 despite advanced cancer [11]. Therefore CA 19-9 has a limited use as a screening tool and the development of new safe, accurate, cost-effective biomarkers is required urgently. Unfortunately, due to the multifactorial nature of the disease and genetic heterogeneity among populations this task has proved more difficult than theorised.
Biomarker discovery can target either DNA, RNA or specific proteins. Specimens suggested for testing include pancreatic tissue, pancreatic juice or body fluids such as serum/plasma. At present there are no good DNA based biomarkers identified for pancreatic cancer. Gene expression profiling studies have identified certain genes of potential diagnostic significance for pancreatic cancer, although there is no clinical application at present. Proteomics involves the study of the complete protein complement. In contrast with the human genome, the proteome is dynamic, in a state of constant flux due to various modifications and regulation. Analysis, therefore, of the proteome not only provides information relating to a mutated gene, but also the extent of its expression at a specific time point. In order to identify an appropriate biomarker for cancer diagnosis, the protein product of an overexpressed gene should be a secreted protein and not expressed in benign pancreatic conditions and other organs. The aim of cancer proteomics commences with the comparison of proteomes from diseased and control samples in large scale to identify differentially expressed proteins (up- or down-regulated) for further quantification and identification. The ideal specimen for such assessment is serum due to its ease of repeated collection and availability. Traditionally proteomic studies have been based around 2-dimensional gel electrophoresis with mass spectrometry. This involves the identification of differentially abundant protein spots which can be excised and digested and potentially identified. Bloomston conducted a large scale study on 62 patients (32 with pancreatic cancer and 30 controls) using 2-dimensional gel electrophoresis. A total of 154 differentially expressed protein spots which could distinguish between the two groups were identified, with nine showing accurate discrimination [12]. Unfortunately this technique is extremely labour intensive and does not work well for small proteins, limiting its use to low throughput studies and, therefore, restricting its application in biomarker discovery for screening purposes [10, 13].
There have been significant developments in mass spectrometry technology in the last two decades. Using these advancements an alternative approach to biomarker discovery has been developed in the form of proteomic pattern analysis. This method utilises the patterns of signals created within a mass spectrum to distinguish between groups of samples such as disease and control. One such method is surface enhanced laser desorption ionization time of flight mass spectrometry (SELDI TOF MS). In SELDI TOF MS, the protein mixture or analyte is spotted on a plate or ProteinChip® (Ciphergen Biosystems Inc., Fremont, CA, USA) where the surface has been modified with a chosen chemical functionality. Due to the chemical functionality, some of the different proteins present in the sample bind to the surface, while the others are not bound and are therefore removed by washing. After washing the spotted sample, a matrix material is applied to the surface and allowed to crystallize with the bound sample proteins. The selective binding acts as a further separation step as only a subset of proteins in the analyte bind to the surface. There are several different available SELDI ‘Chips’ available with different chemical functionality. Surfaces can also be ‘functionalised’ with antibodies, other proteins, or DNA. The chip surface is then pulsed with a UV laser and the bound proteins ionize. The ions generated are accelerated in a high vacuum along a time of flight tube to a detector. Since their time of flight before they reach the detector is a function of their molecular mass, the detector is able to separate out the individual ions generated. This technique has shown promising results in the identification of serum biomarkers for detecting breast, ovarian and prostate cancers [14, 15, 16]. SELDI has numerous advantages over other techniques to date showing a high tolerance to impurities and very low sample requirements. Additionally with prefractioning of the samples detection of low abundance proteins can be improved. A number of biomarkers can be identified and compared using bioinformatic tools allowing high throughput analysis [13]. Unfortunately, there are certain limitations to this technique in the analysis of complex molecules as different molecules do not absorb the energy from the UV laser in the same manner, resulting in different ionisation characteristics from molecule to molecule. Many molecules are completely transparent to UV light and therefore do not ionise at all. The result is that only molecules which highly absorb the UV energy are accessible. Lower absorbing molecules can only be analysed by increasing the UV laser energy, but this causes molecular fragmentation and pyrolysis, leading to erroneous readings.
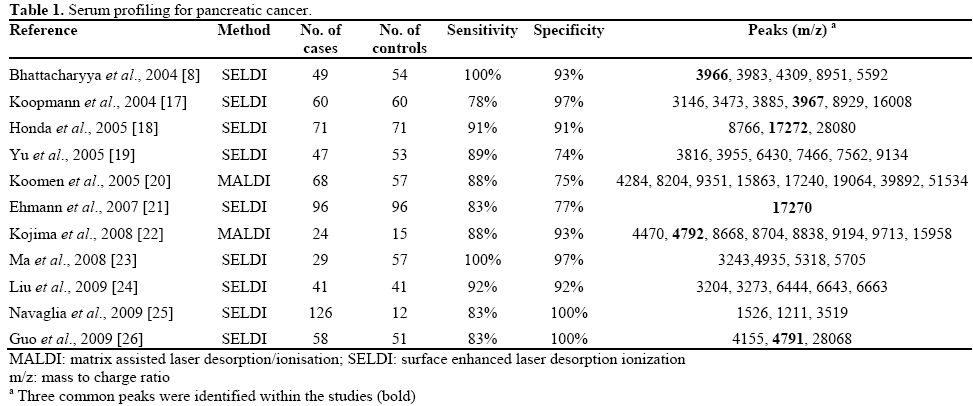
The technique of matrix assisted laser desorption/ ionisation MS (MALDI MS) assists the investigation of complex protein mixtures. It too is often coupled to a time of flight (TOF) detector and is able to provide molecular weight information on the components of protein mixtures. MALDI TOF MS overcomes many of the problems associated with SELDI TOF MS by using a matrix mixed with the analyte. The matrix is usually a highly UV energy absorbing organic acid which has been protonated at acidic pH. The matrix takes over the job of desorption and ionisation of the UV laser energy in the analyte, without causing any damage to the protein or peptide molecules. The presence of matrix molecules, usually in vast excess compared to the analyte, also prevents analyte intermolecular association and increases the useful mass range of the analysis considerably. Proteomics studies on pancreatic cancer in the last few years have focused mostly on the SELDI approach. A majority of these studies have used serum as the diagnostic specimen from pancreatic cancer patients, healthy controls and in certain studies patients with benign pancreatic conditions. Table 1 summarises the results to date of serum based studies using either the SELDI or MALDI technique for profiling pancreatic cancer. These studies have shown promising results in distinguishing pancreatic cancer with healthy controls with a sensitivity ranging from 78-100% and specificity from 74-100%. These are certainly better than the current CA 19-9 marker. Koopman et al. reported superior diagnostic performance when combining the serum SELDI markers with CA 19-9 in comparison with CA 19-9 alone. A particular benefit discussed is the ability to diagnose smaller cancers [17]. On reviewing the peaks identified within the spectra, there have been three common peaks identified within the studies (mass to charge ratio; m/z: 3966, 4791, 17272), with some studies identifying members of the apolipoportein group from other peaks. The fact that such studies have independently identified the same proteins shows promise in using such a technique in the future, however with such a variety of peaks identified further independent studies are required for clinical validation.
Whilst the results with this technique using small samples numbers are encouraging, certain pitfalls have also been identified. As mentioned above, serum remains the ideal choice of specimen due to its easy availability. Despite this the analysis of low abundance proteins in serum remains a difficult task, especially as proteins such as albumin, immunoglobulins, transferrin and macroglobulin make up 80% of all serum proteins [10]. Various additional methods including prefractionation of the serum to remove high abundance proteins have also been developed, with the risk of excluding other proteins of interest. The peptides peaks identified from the samples represent the specific protein profile. The hope is to identify these proteins using specific software and the protein database. Unfortunately, to date, only a small proportion of the peaks postulated as significant biomarkers have been identified, with most of them acute phase reactants associated with general disease. This further limits the use of such proteins as valid biomarkers.
Bioinformatics tools have become integral components of this technology. They are necessary for the storage and analysis of large amounts of data, together with the ability to present the results in a recognisable format for pattern recognition. By using training sets of protein spectra created by mass spectrometry, statistical algorithms can be developed that can cluster results and distinguish between healthy and diseased samples based on these patterns. Petricoin et al. using SELDI TOF-MS developed such an algorithm for detecting a specific proteomic pattern in the diagnosis of ovarian cancer with a sensitivity of 100% and specificity of 95% [15]. One area of concern, however, is regarding the presence of chemical noise in the spectra. Low level protein contamination can lead to variation within the spectra. This experimental error can lead to variations in the analysis that may be unrelated to the biological consequences of the cancer in study but processing of the samples [27]. Another area of concern associated with bioinformatics, as described by De Noo et al. is the issue of overfitting. This involves the analysis of larger datasets where discrimination is identified based on over interpretation of data making the studies not reproducible. This problem can be minimised by methods of double cross validation [28].
The area of bias and error within this technique not only involves the methodology of sample preparation and analysis, but also the biological variables within the population groups. It is theorised that such factors such as different diseases, age, gender, race and drug treatment may affect the proteome of subjects introducing further potential error in the analysis. Similarly with the technology evolving other factors such as sample handling and preparation with advancements in algorithms may have an impact on the results. Naturally this will affect the reproducibility and overall validity of these biomarker studies. For example Karsan et al. produced algorithms that were able to discriminate between breast cancer patients and healthy controls with modest success but were very accurate in selecting which of two clinics had prepared the samples and on what day [29]. Another study by Engwegen et al. attempted to replicate earlier studies of renal cell carcinoma patients carried out by Won et al. and Tolson et al. [30, 31, 32]. They were unable to replicate many of the discriminating peaks and instead found others that were not in the earlier studies. To some extent this may have been due to experimental procedure variation but the authors also postulate that the differences could be due to the relatively small sample sizes used in all the studies (50-75) and consequent variation in the composition of the sample population. A similar situation is seen in studies on pancreatic cancer to date.
The use of mass spectrometry based proteomic profiling in studying pancreatic cancer whilst still in its early stages has highlighted some promising results. However, with this certain potential difficulties have also been identified. With advancing technology leading to improved sensitivity, quantification and reproducibility it is hoped this method will aid identifying low abundance proteins to be used as valid biomarkers for pancreatic cancer.
Conflict of interest
The authors have no potential conflict of interest
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics 2008. CA Cancer J Clin 2008; 58:71-96. [PMID 18287387]
- Christein JD, Kendrick ML, Iqbal CW, Nagorney DM, Farnell MB. Distal pancreatectomy for resectable adenocarcinoma of the body and tail of the pancreas. J Gastrointest Surg 2005; 9:922-7. [PMID 16137585]
- Sperti C, Pasquali C, Piccoli A, Pedrazzoli S. Survival after resection for ductal adenocarcinoma of the pancreas. Br J Surg 1996; 83:625-31. [PMID 8689203]
- Yeo CJ, Cameron JL, Sohn TA, Lillemoe KD, Pitt HA, Talamini MA, et al. Six hundred fifty consecutive pancreaticoduodenectomies in the 1990s: pathology, complications and outcomes. Ann Surg 1997; 226:248-57. [PMID 9339931]
- Moossa AR, Gamagani RA. Diagnosis and staging of pancreatic neoplasms. Surg Clin North Am 1995; 75:871-90. [PMID 7660251]
- Hruban RH, van Mansfield AD, Offerhaus GJ, van Weering DH, Allison DC, Goodman SN et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 1993; 143:545-54. [PMID 8342602]
- Hruban RH, Offerhaus GJ, Kern SE, Goggins M, Wilentz RE, Yeo CJ. Tumor-suppressor genes in pancreatic cancer.J Hepatobiliary Pancreat Surg 1998; 5:383-91. [PMID 9931387]
- Bhattacharyya S, Siegel ER, Petersen GM, Chari ST, Suva LJ, Haun RS. Diagnosis of pancreatic cancer using serum proteomic profiling. Neoplasia 2004; 6:674-86. [PMID 15548376]
- Egawa S, Takeda K, Fukuyama S, Motoi F, Sunamura M, Matsuno S. Clinicopathological aspects of small pancreatic cancer. Pancreas 2004; 28:235-40. [PMID 15084963]
- Chen R, Pan S, Brentnall TA, Aebersold R. Proteomic profiling of pancreatic cancer for biomarker discovery. Mol Cell Proteomics 2005; 4:523-33. [PMID 15684406]
- Goggins MG. The molecular diagnosis of pancreatic cancer. In: Von Hoff DD, Evans DB, Hruban RH. Eds. Pancreatic Cancer. 1st ed. Sudbury: Jones and Bartiett Publishers 2005: 251-64.
- Bloomston M, Zhou JX, Rosemary AS, Frankel W, Muro-Cacho CA, Yeatman TJ. Fibrinogen gamma overexpression in pancreatic cancer identified by large-scale proteomic analysis of serum samples. Cancer Res 2006; 66: 2592-9. [PMID 16510577]
- Seibert V, Ebert MPA, Buschmann T. Advances in clinical cancer proteomics: SELDI-Tof-mass spectrometry and biomarker discovery. Brief Funct Genomic Proteomic 2005; 4:16-26. [PMID 15975261]
- Li J, Zhang Z, Rosenzweig J, Wang YY, Chan DW. Proteomics and bioinformatics approaches for identification of serum biomarkers to detect breast cancer. Clin Chem 2002; 48:1296-304. [PMID 12142387]
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002; 359:572-7. [PMID 11867112]
- Banez LL, Prasanna P, Sun L, Ali A, Zou Z, Adam BL, et al. Diagnostic potential of serum proteomic patterns in prostate cancer. J Urol 2003; 170:442-6. [PMID 12853795]
- Koopmann J, Zhang Z, White N, Rosenzweig J, Fedarko N, Jagannath, S et al. Serum diagnosis of pancreatic adenocarcinoma using surface enhanced laser desorption and ionization mass spectrometry. Clin Cancer Res 2004; 10:860-8. [PMID 14871961]
- Honda K, Hayashida Y, Umaki T, Okusaka T, Kosuge T, Kikuchi S, et al. Possible detection of pancreatic cancer by plasma protein profiling. Cancer Res 2005; 65:10613-22. [PMID 16288055]
- Yu Y, Chen S, Wang LS, Chen WL, Guo WJ, Yan H, et al. Prediction of pancreatic cancer by serum biomarkers using surface enhanced laser desorption/ionization-based decision tree classification. Oncology 2005; 68:79-86. [PMID 15864000]
- Koomen JM, Shih LN, Coombes KR, Li D, Xiao LC, Fidler IJ, et al. Plasma protein profiling for diagnosis of pancreatic cancer reveals the presence of host response proteins. Clin Cancer Res 2005; 11:1110-8. [PMID 15709178]
- Ehmann M, Felix K, Hartmann D, Schnolzer M, Nees M, Vorderwulbecke S, et al. Identification of potential markers for the detection of pancreatic cancer through comparative serum protein expression profiling. Pancreas 2007; 34:205-14. [PMID 17312459]
- Kojima K, Asmellash S, Klug CA, Grizzle WE, Mobley JA, Christein JD. Applying proteomic-based biomarker tools for the accurate diagnosis of pancreatic cancer. J Gastrointest Surg 2008; 12:1683-90. [PMID 18709425]
- Ma N, Ge CL, Luan FM, Hu CJ, Li YZ, Liu YF. Establishment of serum protein pattern model for screening pancreatic cancers by SELDI-TOF-MS technique.Zhonghua Wai Ke Za Zhi 2008; 46:932- 5. [PMID 19035154]
- Liu D, Cao L, Yu J, Que R, Jiang W, Zhou Y, et al. Diagnosis of pancreatic adenocarcinoma using protein chip technology. Pancreatology 2009; 9:127-35. [PMID 19077463]
- Navaglia F, Fogar P, Basso D, Greco E, Padoan A, Tonidanel L, et al. Pancreatic cancer biomarkers discovery by surface-enhanced laser desorption and ionization time-of-flight mass spectrometry. Clin Chem Lab Med 2009; 47:713-23. [PMID 19426140]
- Guo J, Wang W, Liao P, Lou W, Ji Y, Zhang C, et al. Identification of serum biomarkers for pancreatic adenocarcinoma by proteomic analysis. Cancer Sci 2009; 100:2292-301. [PMID 19775290]
- Rodland K. Proteomics and cancer diagnosis: the potential of mass spectrometry. Clin Biochem 2004; 37:579-83. [PMID 15234239]
- De Noo ME, Tollenaar RAEM, Deelder AM, Bouwman LH. Current status and prospects of clinical proteomics studies on detection of colorectal cancer: Hopes and fears. World J Gastroenterol 2006; 12:6594-601. [PMID 17075970]
- Karsen A, Beruhard JE, Flibotte S, Gelmon K, Switzer P, Hassell P, et al. Analytical and preanalytical biases in serum proteomic pattern analysis for breast cancer diagnosis. Clin Chem 2005; 51:1525-8. [PMID 15951319]
- Engwegen JY, Mehra N, Haanen JB, Bonfrer JM, Schellens JH, Voest EE, Beijnen JH. Validation of SELDI-TOF MS serum protein profiles for renal cell carcinoma in new populations. Lab Invest 2007; 87:161-72. [PMID 17318195]
- Won Y, Song HJ, Kang TW, Kim JJ, Han BD, Lee SW. Pattern analysis of serum proteome distinguishes renal cell carcinoma from other urologic diseases and healthy persons. Proteomics 2003; 3: 2310-2316. [PMID 14673781]
- Tolson J, Bogumil R, Brunst E, Beck H, Elsner R, Humeny A, et al. Serum protein profiling by SELDI mass spectrometry: detection of multiple variants of serum amyloid alpha in renal cancer patients. Lab Invest 2004; 84:845-56. [PMID 15107802]

