Esmaiel IF Saad1 and Idress Hamad Attitalla2*
1Department of Microbiology, Omar Al-Mukhtar University, Al Bayda, Libya
2Department of Botany, Omar Al-Mukhtar University, Al Bayda, Libya
*Corresponding Author:
Idress Hamad Attitalla
Faculty of Science
Department of Botany
Omar Al-Mukhtar University
Box919, Al-Bayda, Libya
Tel: +218910540392
E-mail: idressattitalla2004@yahoo.com
Received Date: 13 November 2017; Accepted Date: 27 November 2017; Published Date: 04 December 2017
Citation: Saad EIF (2017) Molecular Dynamic Simulation and an Inhibitor Prediction of PI3K (P110 α) Subunit Mutant Protein Structured Model as a Potential Drug Target for Cancer. Vol. 1 No. 1: 7.
Copyright: © 2017 Saad EIF. This is an open-access article distributed under the terms of the creative Commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Keywords
PI3K (P110 α) subunit; Modeller; Molecular docking; Molecular dynamic
Introduction
For last many millennium Cancers have been one of the most dreadful and devastating disease. It is characterized by uncontrolled cell growth and the spread of these abnormal cells [1]. Over time, carcinogens induce individual cells to acquire genetic and epigenetic changes in signaling pathways that regulate their growth and proliferation [2]. It has been previously reported that PI3K (Phosphoinositid-3-Kinase) is most of a proto oncogene, which have an essential role in controlling the activity of several crucial cells signaling pathways that enhance the stimulation of cellular replication and cell proliferation and to inhibit growth and apoptosis [3,4]. It was reported The PI3K protein family classified according to their sequence, domain structure and mode of regulation, To three groups, i.e., Class I PI3K, Class II PI3K and class III PI3K [5]. Class I PI3K has three subunit classes, viz., p110 α, p110 β and p110γ which plays an important role in cancer. Several studies document the fact PI3K (P110α) subunit protein is mostly responsible to be frequently mutated protein in several types of cancers such as breast (27%), endometrial (23%), urinary tract (17%), colorectal (14%) and ovarian (8%) cancers [4,6-8]. Moreover, the mutations in PI3K (P110α) subunit lead to constitutive activation of downstream pathways resulting in the altered regulation of cellular proliferation and malignant transformation which makes it a very attractive drug target for cancer biology, Development of multidrug resistance and of the molecular simulation approaches which strongly justifies the need of the identification of drug targets in cancer, which is a major challenge in modern medicine. When we consider through treatment for cancer currently, they depend on the type and stages of cancer development. Chemotherapy, surgery, radiation therapy, and hormonal therapy are the various treatments that currently take place, But these types of treatments except for the target based, cannot distinguish between normal and tumorous cells [9]. Therefore, we aim to concentrate on three assumptions to postulate the novel drug target for cancer: (1) to predict the structural model of PI3K (P110 α) subunit mutant protein, (2) to investigate structural analysis of PI3K (P110 α) subunit mutant protein using molecular dynamics simulation studies, and (3) to perform virtual screening and docking to identify inhibitor molecules against the suitable ligand. Many therapeutic agents currently being evaluated have multiple targets and their antitumor effects may not be due to specific mutated PI3K (P110α) subunit inhibition. Hence, the present investigation was carried out to utilize PI3K (P110α) subunit as a canonically relevant anticancer drug target in cancer therapy.
Materials and Methods
Selection of the drug target
A mutation in PI3K (P110α) subunit protein at codon 542, 545, and 1047 the most common mutational events in human carcinogenesis and has been found in a variety of human cancers, with the frequent replacement of the amino acids, Glu by Lys at codon 542 and Glu by Lys or Gln at codon 545 and His by Age or Leu at codon 1047 [10,11]. Moreover, the mutations in PI3K (P110α) subunit lead to constitutive activation of downstream pathways resulting in the altered regulation of cellular proliferation and malignant transformation which makes it a very attractive drug target for cancer biology. Therefore, in the present study, we have selected PI3K (P110α) subunit as the most promising drug target for the evaluation of the molecular dynamics simulations.
Model generation of the selected drug target
For putative model generation, we retrieved the PI3K (P110 α) subunit (target) amino acid sequence from the UniProt database (https://www.uniprot.org) Furthermore, the Protein query sequence (P42336) in fasta format was downloaded from UniProt (https://www.uniprot.org/uniprot/) databases. The 3D structures of all wild type and mutated protein were predicted using Modeller 9.17 software [12]. All possible (hotspot) mutations reported for codons 542, 545 and 1047 of the PI3K (P110 α) subunit proto-oncogene were retrieved through a literature survey [5,7,8,13]. The structural models were further evaluated using Structural Analysis and Verification Server’s PROCHECK tool which checks the stereochemical quality of a protein structure utilizing Ramachandran plot [14]. The best models from each individual protein (wild type as well as mutated) were taken under consideration for further analysis. A prediction of active site of all mutated PI3K (P110 α) subunit conformations was performed with putative active sites with sphere software, PASS [15].
Molecular dynamics simulation study of the predicted model
In our study, we performed molecular dynamics (MD) simulations of modeled PI3K (P110 α) subunit protein using GROMACS 4.0.6 software package [16] with GROMOS 96 force fields [17] and the flexible SPC water model. The initial structure was immersed in a periodic water box of cubic shape (0.5 nm thick). Electrostatic energy was calculated using the particle mesh Ewald method, which permits the use of the Ewald summation at a computational cost comparable to that of a simple truncation method of 10A or less [18]. We retained the cutoff distance as 1.0 nm for the calculation of the coulomb and Van der Waal’s interaction, respectively. After energy minimization using a steepest descent for 1000 steps, the system was subjected to equilibration at 300K and normal pressure for 100 ps under the conditions of position restraints for heavy atoms. We subsequently applied LINCS [19] constraints for all bonds, keeping the whole protein molecule fixed and allowing only the water molecule to move to equilibrate with respect to the protein structure. The system was coupled to the external bath by the Berendsen pressure and temperature coupling [20]. The final MD calculations were performed for 10 ns under the same conditions except that the position restraints were removed. The results were analyzed using the standard software provided by the GROMACS package. An average structure was further refined using the steepest descent energy minimization.
Virtual screening
For further utilized for virtual screening. A library of anticancer drugs was prepared on the basis of literature retrieved from different sources’ viz., PubChem (https://pubchem.ncbi.nlm.nih.gov/) and ChemSpider of the Royal Society of Chemistry (https://www.chemspider.com/). In silico virtual screening was carried out using PyRx [21]. Software to rank all the library molecules under study, according to their affinity towards the active site of PI3K (P110 α) subunit mutated protein conformations. All the library molecules (i.e., top ten putative library molecules) were ranked according to their affinity towards the mutated PI3K (P110 α) subunit conformations.
Results and Discussion
Prediction and validated modeled structure of the PI3K (P110 α) subunit protein
The structure of PI3K (P110 α) subunit protein wild type and five mutated models have determined by using homology modeling protocol, By using Modeller 9.17 software. Firstly BLASTP search was performed against PDB with default parameters to find suitable templates for homology modeling. Based on the maximum identity with high score and lower evalue (Template) were used as the template for homology modeling. The final stable structure of PI3K (P110 α) subunit protein is shown in Figure 1. The mutations were selected according to the earlier literature [10,22-27]. For each protein three models were generated. The selected protein models for wild type and another three mutated codon 542, 545 and 1047 PI3K (P110 α) subunit conformations. The mutations occurring in codon 542, 545 and 1047 due to the amino acid changes are clearly shown in Figure 1. The models were analyzed online by submitting to NIH MBI Laboratory for Structural Genomics and Proteomics’ SAVES server. Validity reports generated by PROCHECK and Verfiy_3D judged accuracy of the protein models. A comparison of the results obtained from the above mentioned validation tools, showed that one of the models generated by Modeller is more acceptable in comparison to the others. So, one of every three most valid model was selected for each protein model to use for further studies. It was found that the phi/psi angles of 85.1 to 87.3% of the residues fell in the most favored regions, 9.1 to 11.7% of the residues fell in the additional allowed regions, 2.0 to 2.4% fell in the generously allowed regions, and 0.8 to 1.7 of the residues fell in the disallowed regions in all PI3K (P110α) subunit wild type and all other proteins. A score of >50% in the most favored regions is acceptable for a reasonable protein model and the score obtained more than 80% indicate the quality of all selected PI3K (P110 α) subunit protein models. The identification of the active site in the mutated protein models was followed by the screening and identification of potential inhibitor molecules targeting active site toward mutant codon 545, 542 of PI3K (P110 α) subunit protein.
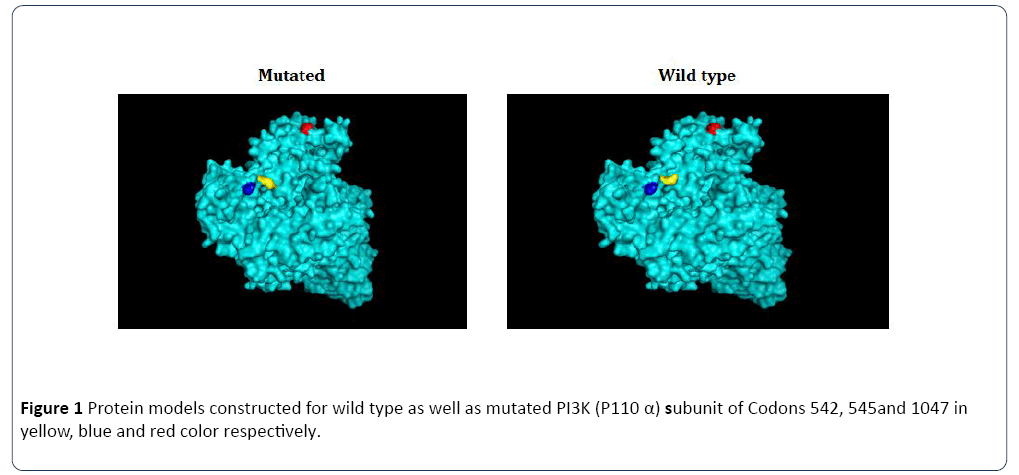
Figure 1: Protein models constructed for wild type as well as mutated PI3K (P110 α) subunit of Codons 542, 545and 1047 in yellow, blue and red color respectively.
Molecular dynamic simulation of PI3K (P110 α) subunit mutant protein
In this study, the predicted model of PI3K (P110 α) subunit mutant protein has shown better accuracy of the structure as revealed by the virtual screening. It has been earlier described that the virtual screened model should have the stable molecular dynamic behavior. We evaluated the virtual screened model of PI3K (P110 α) subunit protein using the molecular dynamic stability criteria by performing the molecular dynamic simulation study using GROMACS 4.0.6 software package. In this case, a molecular dynamics simulation study revealed a consistent value of the energy of the molecule throughout the time period of simulation, which is an indication of strong basis of the fact that the molecule has a stable structure as required for the virtual screening and drug designing processes (Figures 1-4). The computed equilibrium configuration of the protein models showed a gradual decrease in the potential energy (-6.97 to -7.02), temperature (300/ K), pressure mean between (-200 to 100) and density was between (1020 to 1025) of protein molecules with the increase in time (0 to 100 picoseconds) and reached to an equilibrium (Figures 1-4). Energy minimization was performed in order to eliminate or reduce potential geometric problems in the protein structures such as bond distances, bond angles and torsion angles. We further evaluated the radius of gyration parameter (Rg) is a measure often used to describe the compactness of a given protein. It was observed that initially the radius of gyration was increasing, The computed data on Rg for all protein models were in the range of 3.3 to 4.0 nanometers against a function of time from 0 to 100 picoseconds (Figure 5). And finally, become almost constant for the rest of the duration of the simulation (Figure 5). This observation suggests and is in line with the argument that the PI3K (P110 α) subunit mutant protein model has a compact structure, which provides the required stability to the molecule and supports our previous molecular dynamic simulation results. Root mean square deviation (RMSD) was evaluated during the simulation, and it was observed that it was increasing in the beginning, but after 2000 ps it became almost constant for the rest of the duration of the simulation, which suggests that the hypothesized PI3K (P110 α) subunit protein model has a lesser RMSD for the PI3K (P110 α) subunit protein backbone and has less flexibility, indicating the stable dynamic behavior structure of PI3K (P110 α) subunit protein (Figure 6). It is evident from the data that the RSMD values initiated around 0.15 to 0.2 nanometers, gradually increased up to 0.2 nanometers at 100 picoseconds and then leveled off (Figure 6). This indicates that after an initial increase in the magnitude of protein atoms fluctuation, the protein reached an equilibrium state characterized by the RMSD profile.
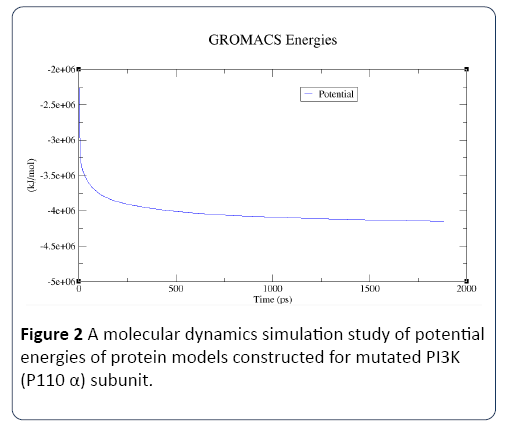
Figure 2: A molecular dynamics simulation study of potential energies of protein models constructed for mutated PI3K (P110 α) subunit.
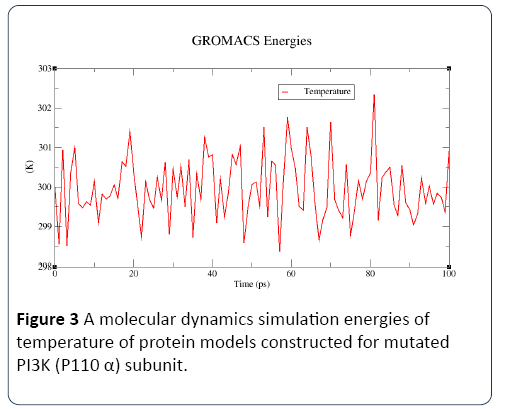
Figure 3: A molecular dynamics simulation energies of temperature of protein models constructed for mutated PI3K (P110 α) subunit.
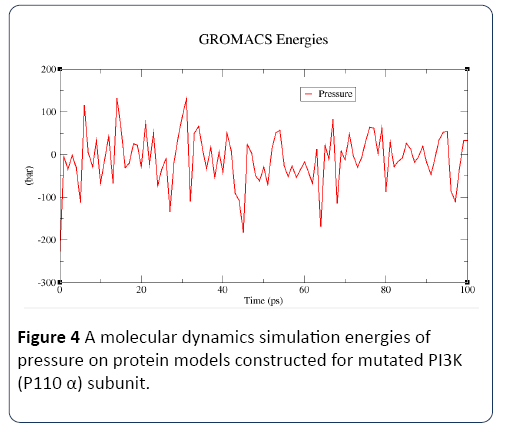
Figure 4: A molecular dynamics simulation energies of pressure on protein models constructed for mutated PI3K (P110 α) subunit.
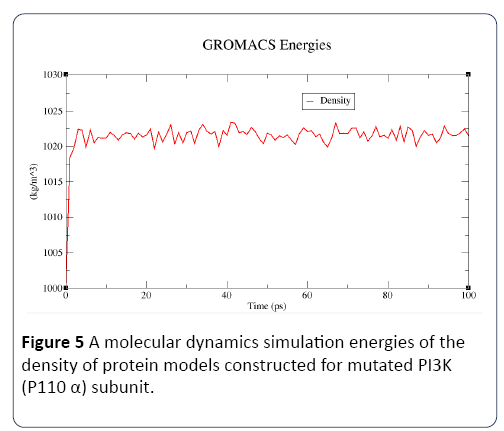
Figure 5: A molecular dynamics simulation energies of the density of protein models constructed for mutated PI3K (P110 α) subunit.
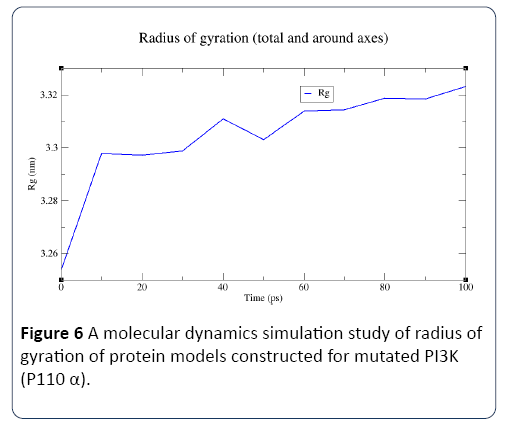
Figure 6: A molecular dynamics simulation study of radius of gyration of protein models constructed for mutated PI3K (P110 α).
The energy plot and RMSD plot showed a substantial increase in their respective values when the time variable was increased from 10 to 100 picoseconds. The Rg plot clearly showed the increase of stability with increase in time. Hence, the 100 picoseconds structural statistics were taken into consideration and the data obtained reveal that protein models have shown stability during molecular dynamic simulations (Figure 7). The present investigation on molecular dynamic simulation was in agreement with different protein simulations using GROMACS simulation [28-34]. Which strongly suggests that PI3K (P110 α) subunit protein model has an accurate and stable structure and can be used for the virtual screening.
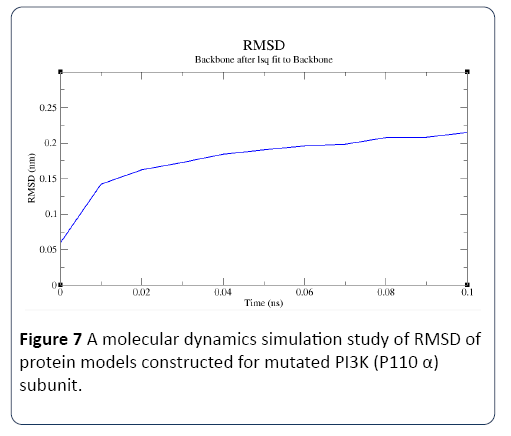
Figure 7: A molecular dynamics simulation study of RMSD of protein models constructed for mutated PI3K (P110 α) subunit.
Virtual screening
Towards finding suitable inhibitor(s), ten probable inhibitor molecules with lower docking energies were chosen individually for PI3K (P110 α) subunit mutations. Each inhibitor molecule was viewed and the moieties having lower docking energies were chosen as the possible PI3K (P110 α) subunit inhibitor molecules and their ranking according to the lowest docking energies. We observed that four ligands showed the best derivatives of pyrazolo pyrimidine, 5a: -21. 44 kcal/mol; pyrazolo pyrimidine, 10: -17.67 kcal/mol; pyrimidine, 25: -16.44 kcal/mol; Sorafenib,: -13.4 kcal/mol. The computational approach that docks small molecules into the structures of macromolecular targets and score their potential complementary to binding sites are now widely used in hit identification and lead optimization [35,36]. The present results in identification of potential inhibitor molecules based on the lowest docking energies are in agreement with Garcia- Echeverria and Sellers et al. [30,37-40] in their Docking studies of PI3K (P110 α) subunit with various compounds revealed against selected active site which was the best to be targeted by all hits and showed a good docking score just similar to our docking score.
Conclusion
From this in silico study and previously reported experimental data in the literature, Based on intrinsic dynamics, structural stability and the improved relaxation of modeled protein, the energy of the structure, the radius of gyration, RMSD and RMS fluctuation, the protein modeled were in stable conformation. And we conclude that the four ligand molecule, are the effective chemical molecule and having the highest binding affinity towards mutant PI3K (P110 α) subunit protein, which will in turn arrest the process of cell growth and proliferation of the cancerous cells.
Acknowledgment
The authors would like to acknowledge the supporting and the facilities provided by Department of computer science, faculty of Science Omar Al Mukhtar University, Bayda, Libya.
References
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57-70.
- Heeg S, Doebele M, von Werder A, Opitz OG (2006) In vitro transformation models: modeling human cancer. Cell Cycle 5: 630-634.
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655-1657.
- Zunder ER, Knight ZA, Houseman BT, Apsel B, Shokat KM (2008) Discovery of drug-resistant and drug-sensitizing mutations in the oncogenic PI3K isoform p110α. Cancer Cell 14: 180-192.
- Miller TW, Rexer BN, Garrett JT, Arteaga CL (2011) Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res 13: 224-235.
- Maira SM, Voliva C, Garcia-Echeverria C (2008) Class IA PI3 Kinase: From their biological implication in human cancers to drug discovery. Expert Opin Ther Targets 12: 223–238.
- Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7: 606–619.
- Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27: 5497–5510.
- Vincent S, Djulbegovic B (2005) Oncology treatment recommendations can be supported only by 1-2% of high quality published evidence. Cancer Treat Rev 31: 319-322.
- Samuels Y, Velculescu VE (2004) Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 3: 1221–1224.
- Vogt PK, Hart JR, Gymnopoulos M, Jiang H, Kang S, et al. (2011) Phosphatidylinositol 3-kinase (PI3K): The oncoprotein. Curr Top Microbiol Immunol 347: 79–104.
- Sali A, Potterton L, Yuan F, Vlijmen VH, Karplus M (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23: 318-326.
- Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, et al. (2011) PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mole Cancer Ther 10: 558-565.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK - a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283-291.
- Brady GP, Stouten PF (2000) Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des 14: 383-401.
- Hess B, Kutzner C, Spoel DVD, Lindahl E (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435-447.
- Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF (2004) A Biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 25: 1656-1676.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: A linear constraint solver for molecular simulations. J Comput Chem 18: 1463-1472.
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, et al. (1995) A smooth particle mesh Ewald method. J Chem Phys 103: 8577-8593.
- Berendsen HJC, Postma JPM, Van Gunsteren WF, Dinola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81: 3684-3690.
- Jacob RB, Andersen T, McDougal OM (2012) Accessible high-throughput virtual screening molecular docking software for students and educators. PLoS Comput Biol 8: e1002499.
- Lee JW, Soung YH, Kim SY, Lee HW, Park WS (2005) PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 24: 1477-1480.
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, et al. (2005) Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 65: 10992-11000.
- Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, et al. (2005) The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. PNAS 102: 18443-18448.
- Bader AG, Kang S, Vogt PK (2006) Cancer-specific mutations in PIK3CA are oncogenic in vivo. PNAS 103: 1475–1479.
- Samuels Y, Ericson K (2006) Oncogenic PI3K and its role in cancer. Curr Opin Oncol 18: 77–82.
- Zhao L, Vogt PK (2008) Class I PI3K in oncogenic cellular transformation. Oncogene 27: 5486–5496.
- Saad EIF (2017) Drug design for cancer-causing pi3k (p110 α) subunit mutant protein. (The Role of Chemistry in Applied Research and Sustainable Development), 2nd Libyan Conference on Chemistry and Its Applications, LCCA-2. pp: 141–146.
- Sabbah DA, Simms NA, Brattain MG, Vennerstrom JL, Zhong H (2012) Biological evaluation and docking studies of recently identified inhibitors of phosphoinositide-3-kinases. Bioorg Med Chem Lett 22: 876–880.
- Sabbah DA, Vennerstrom JL, Zhong HA (2012) Binding selectivity studies of phosphoinositide 3-kinases using free energy calculations. J Chem Inf Model 52: 3213-3224.
- Singh S, Sablok G, Farmer R, Singh AK, Gautam B, et al. (2013) Molecular dynamic simulation and inhibitor prediction of cysteine synthase structured model as a potential drug target for trichomoniasis. Biomed Res Int, pp: 1-15.
- Gkeka P, Evangelidis T, Pavlaki M, Lazani V, Christoforidis S, et al. (2014) Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput Biol 10: e1003895.
- Li T, Wang G (2014) Computer-aided targeting of the PI3K/Akt/mTOR pathway: Toxicity reduction and therapeutic opportunities. Int J Mol Sci 15: 18856–18891.
- Zhang Y (2015) Understanding the impact of Fc glycosylation on its conformational changes by molecular dynamics simulations and bioinformatics. Mole BioSystems 11: 3415-3424.
- Gao Y, Ma Y, Yang G, Yiping LY (2016) Molecular dynamics simulations to investigate the binding mode of the natural product liphagal with phosphoinositide 3-kinase. Molecules 21: 857-868.
- Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat Rev Drug Discov 3: 935-949.
- Kroemer RT (2007) Structure based drug design: docking and scoring. Curr Protein Pept Sci 8: 312-328.
- Garcia-Echeverria C, Sellers WR (2008) Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 27: 5511–5526.
- Sujatha S, Silja S (2011) Finding the potential Inhibitors of Phosphoinositide 3-kinase as a anti-cancerous drug target. J Biotechnol 3: 177-185.
- Chaudhary B, Singh S (2012) Molecular docking studies on pyrazolopyrimidine and their derivatives as human phosphoinositide 3-kinase inhibitors. Int J Adv Bioinfo Comp Biolo 1: 1-11.