Keywords
CNPC; PES; NBO; NLO; HOMO-LUMO
Introduction
Organic materials are attractive due to their optical properties, electronics, and integrated photonics [1-3]. Organic molecules with electron deficient pyrimidine ring tend to act as electron acceptor and are very effectively used in Organic light emitting diodes (OLEDs) [4]. The higher light harvesting efficiency achieved by pyrimidine adopted porphyrin sensitizers show more advantage in oxidized dyes. New organic dyes with pyrimidine-2-carboxylic acid forms coordination bond with TiO2 improves the interaction between the anchor and semiconductor [5]. Pyrimidine show considerable efforts in the development of bipolar materials to overcome the unipolar character of the organic materials [6]. Recent work from Lin et al., reported that pyrimidine used as π-conjugated spacer in organic photosensitizers in dye sensitized solar cells (DSSCs) [7]. The electron withdrawing character of pyrimidine chromophore exhibits the white photoluminescence in both liquid and solid state [8]. Moreover, pyrimidine based iridium complex exhibit external quantum efficiency up to 28.6% due to high photoluminescence quantum yield determines the excellent device performance and high efficiency [9].
In this paper, we report the synthesis, spectral and nonlinear optical investigation of ethyl-4-(4-chloro-3-nitrophenyl)-6- methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (CNPC). Here the molecular structure and electronic structural properties of the title molecule were studied using experimental and also with theoretical approach. For unambiguous vibrational spectral assignments precisely potential energy distribution have been performed and related with recorded FT-IR and FTRaman spectra, respectively. This work also covers the molecular electrostatic potential mapped surface and energy gap analysis along with global reactivity descriptors. The charge transfer interactions occur in the CNPC molecule and stabilization arising from the donor-acceptors interactions are examined using Natural bond orbital analysis. The first hyperpolarizability (β) components examining the nonlinear response of the title molecule. The NMR chemical shifts calculated using Gauge-independent atomic orbitals and experimental chemical shifts were also analyzed in the present work.
Experimental
Synthesis of ethyl 4-(4-chloro-3-nitrophenyl)-6- methyl-2-oxo-1,2,3,4-tetrahydro pyrimidine-5- carboxylate
4-Chloro-3-nitrobenzaldehyde (1.84 ml, 0.01 mmol) and urea (1.8 g, 0.03 mmol) was added to an ethanolic solution of ethyl acetoacetate (1.34 ml, 0.01 mmol). To the mixture CeCl3.7H2O (0.465 g, 25%) was added and stirred well. Then, the reaction mixture was refluxed at 90°C for 2-3 hours and the completion of the reaction was monitored by thin layer chromatography. After completion, the reaction mixture was poured onto crushed ice and stirred up to 5-10 minutes. The solid product was separated, filtered under suction, washed with ice-cold water and then recrystallized from absolute ethanol. The synthesis of CNPC molecule is shown in Scheme 1. Melting point=185˚C; Yield=87%.
Scheme 1 The reaction scheme of synthesis of CNPC molecule.
Computational Details
The quantum chemical calculations of CNPC was performed using the B3LYP level of theory supplemented with 6-31G(d,p) basis set, using Gaussian 03 program package invoking geometry optimization [10]. Initial geometry generated from geometrical parameters was minimized without any constraint in the potential energy surface at DFT level. The optimized minimum structure parameters were used in the vibrational wavenumber calculations at the DFT level to characterize all stationary points as minima. The harmonic vibrational wavenumber calculations resulting in IR and Raman intensities and Raman depolarization ratios. The vibrational modes were assigned based on potential energy distribution analysis (PED) using VEDA4 program [11]. The Raman activities were transformed into Raman intensities using Raint program [12] by the expression:
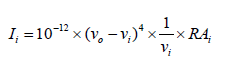
Where Ii is the Raman intensity, Ai is the Raman scattering activities, νi is the wavenumber of the normal modes and ν0 denotes the wavenumber of the excitation laser [13].
Results and Discussion
Molecular conformational analysis
In order to investigate the stable conformer, potential energy surface scan was performed to CNPC molecule. In this PES scan process, the internal redundant coordinate of the dihedral angle D(C11‒C13‒C16‒C20) chosen for the conformational flexibility within the molecule. During this scan the geometrical parameters was relaxed, while the D(C11‒C13‒C16‒C20) torsional angle raised from 0˚ to 360˚ rotation by a step of 10˚ interval. The potential energy surface scan curve and optimized structure of CNPC is shown in Figure 1. From the results, there are three maximum energy conformers (0˚, 170˚ and 360˚) and two minimum conformers (100˚ and 270˚) were identified. The most stable conformer was identified at 270˚ rotation with the relative energy -1542.44631 Hartree. Hence this structure is the global Rotation (°) Relative energy minimum conformer and is used for the further investigations. The various possible conformers of CNPC during PES scan were shown in Table 1.
| Rotation (°) |
Relative energy
(Hartree) |
Rotation (°) |
Relative energy
(Hartree) |
| 0 |
-1542.4 |
190 |
-1542.4 |
| 10 |
-1542.4 |
200 |
-1542.4 |
| 20 |
-1542.4 |
210 |
-1542.4 |
| 30 |
-1542.4 |
220 |
-1542.4 |
| 40 |
-1542.4 |
230 |
-1542.4 |
| 50 |
-1542.4 |
240 |
-1542.4 |
| 60 |
-1542.4 |
250 |
-1542.4 |
| 70 |
-1542.4 |
260 |
-1542.4 |
| 80 |
-1542.4 |
270 |
-1542.4 |
| 90 |
-1542.4 |
280 |
-1542.4 |
| 100 |
-1542.4 |
290 |
-1542.4 |
| 110 |
-1542.4 |
300 |
-1542.4 |
| 120 |
-1542.4 |
310 |
-1542.4 |
| 130 |
-1542.4 |
320 |
-1542.4 |
| 140 |
-1542.4 |
330 |
-1542.4 |
| 150 |
-1542.4 |
340 |
-1542.4 |
| 160 |
-1542.4 |
350 |
-1542.4 |
| 170 |
-1542.4 |
360 |
-1542.4 |
Table 1: The various possible conformers of CNPC.
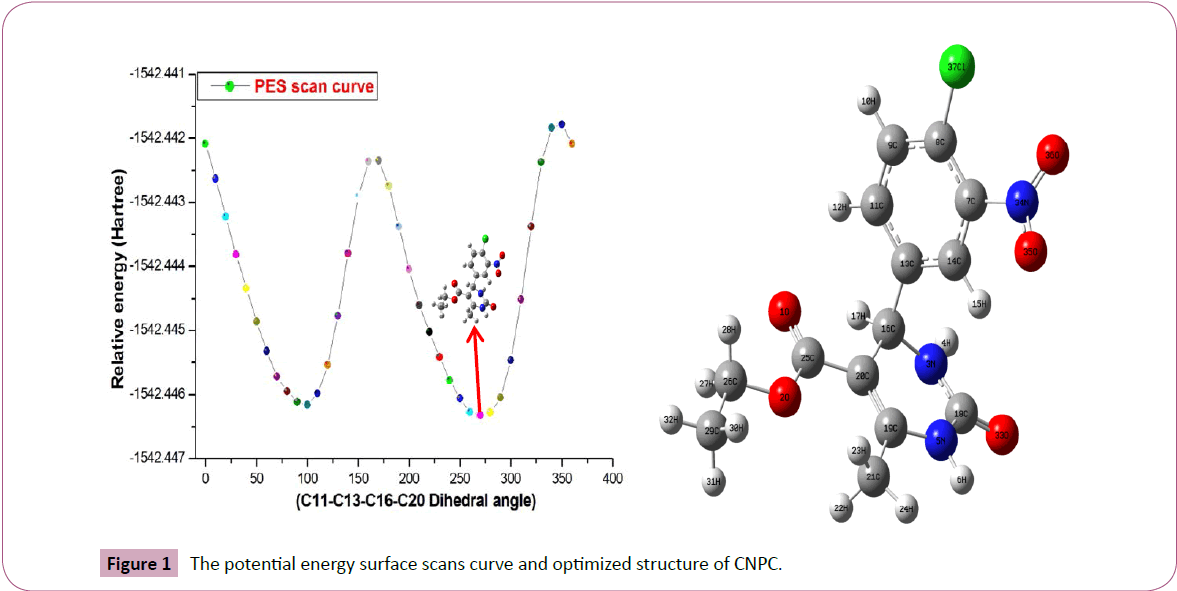
Figure 1: The potential energy surface scans curve and optimized structure of CNPC.
Vibrational Assignments
The detailed description of vibrational assignments of CNPC along with calculated IR/Raman intensities and potential energy distributions of the vibrations are listed in Table 2. For visual comparison, the recorded and simulated FT-IR and FT-Raman spectra of CNPC are shown in Figures 2 and 3, respectively. The un-scaled wavenumber obtained from DFT method, overestimate the observed wavenumber. To bring the theoretical wavenumber closer to the observed wavenumber, a selective scaling procedure was employed. The title molecule have 37 atoms, hence it gives 105 normal modes of vibrations, 36 stretching, 71 in-plane bending and 34 out-of-plane bending vibrations.
| Mode No. |
Calculated wavenumber |
Observed wavenumber |
Intensity |
PED ≥10% |
| Unscaled |
Scaled |
FT-IR |
FT-Raman |
IR |
Raman |
| 1 |
3620 |
3501 |
3354 |
|
6.84 |
1.96 |
νN3H4(99) |
| 2 |
3210 |
3104 |
3109 |
|
0.18 |
2.14 |
νC14H15(95) |
| 3 |
3194 |
3089 |
|
3070 |
0.04 |
1.59 |
νC9H10(96)+ νC11H12(98) |
| 4 |
3078 |
2976 |
2966 |
|
0.67 |
2.48 |
νC16H17(99) |
| 5 |
3036 |
2936 |
2933 |
2934 |
2.24 |
8.36 |
νC29H30(99)+ νC29H31(98)+ νC29H32(98) |
| 6 |
1789 |
1730 |
1699 |
1679 |
100.00 |
6.75 |
νO33C18(69)+ νN3C18(34) |
| 7 |
1715 |
1659 |
1639 |
1635 |
56.86 |
12.96 |
νO1C25(85) |
| 8 |
1663 |
1608 |
1593 |
1613 |
28.76 |
13.15 |
νC19C20(66) |
| 9 |
1639 |
1585 |
|
1593 |
6.43 |
12.49 |
νC9C11(53)+ νC14C7(55) |
| 10 |
1607 |
1554 |
|
|
9.26 |
3.98 |
νO36N34(80)+ νC13C14(58)+ νC7C8(43) |
| 11 |
1591 |
1538 |
1500 |
1503 |
27.45 |
5.59 |
νO36N34(80)+ νO35N34(71)+ νC7C8(43)+βC8C9C11(49) |
| 12 |
1505 |
1455 |
1452 |
|
5.23 |
0.28 |
βH10C9C8(75)+ βH12C11C13(68)+ βH15C14C13(74)+βC7C8C9(25) |
| 13 |
1462 |
1414 |
|
1416 |
15.14 |
4.10 |
βH4N3C16(48)+ τC16C13C20H17(72) |
| 14 |
1422 |
1375 |
1377 |
1375 |
3.70 |
1.97 |
βH22C21H24(66)+ βH22C21H23(74) |
| 15 |
1358 |
1313 |
1323 |
|
6.66 |
1.25 |
νC14C7(55)+ βH17C16C13(63) |
| 16 |
1339 |
1295 |
1280 |
1275 |
19.93 |
3.45 |
νC25C20(18)+ τC26H27C29H28(84) |
| 17 |
1277 |
1235 |
|
1229 |
4.48 |
1.18 |
νN5C19(42)+ βH6N5C19(60) |
| 18 |
1259 |
1217 |
1219 |
1205 |
67.71 |
4.59 |
νN3C18(34)+ τC16C13C20H17(72) |
| 19 |
1214 |
1173 |
1165 |
1168 |
1.12 |
13.48 |
νC8C9(49)+ βC11C13C14(30)+ νC13C16(34)+ βH12C11C13(68) |
| 20 |
1176 |
1137 |
|
1157 |
0.44 |
0.25 |
βH30C29C26(83)+ τC26C29O2H27(87) |
| 21 |
1145 |
1107 |
|
1108 |
0.83 |
1.03 |
νC21C19(15)+ νN3C16(36)+ νC16C20(23) |
| 22 |
1123 |
1086 |
1089 |
1089 |
6.16 |
1.77 |
τC29H30C26H31(65) |
| 23 |
1061 |
1026 |
|
1023 |
9.27 |
7.51 |
νC37l8(48)+ βC8C9C11(49) + βC14C7C8(45)+ βC7C8C9(25) |
| 24 |
1054 |
1019 |
1016 |
|
0.72 |
0.42 |
βH23C21H24(82)+ τC21H24C19H22(82) |
| 25 |
1035 |
1001 |
|
1003 |
0.54 |
2.40 |
νC29C26(64)+ νO2C26(67)+ τC21H22C19H23(46) |
| 26 |
985 |
953 |
956 |
963 |
0.03 |
0.10 |
ÓH10C9C11H12(78)+ ÓH12C11C13C16(88) |
| 27 |
802 |
776 |
|
777 |
0.83 |
2.13 |
βC16N3C18(11)+ τO1C20O2C25(63) |
| 28 |
785 |
759 |
759 |
|
1.36 |
2.19 |
τO36C7O35N34(60)+ τO1C20O2C25(63) |
| 29 |
758 |
733 |
|
723 |
5.65 |
2.34 |
τO33N3N5C18(86) |
| 30 |
653 |
631 |
638 |
|
2.16 |
3.56 |
βO1C25O2(40) |
| 31 |
626 |
605 |
|
595 |
6.11 |
1.55 |
ÓC20C16C19N5(43)+ ÓC21C19C20C25(47)+ τC25C16C19C20(32) |
| 32 |
581 |
562 |
563 |
|
8.64 |
1.92 |
βO35N34C7(26)+ ÓH4N3C18N5(77)+ ÓH6N5C19C21(79) |
| 33 |
517 |
500 |
499 |
|
0.10 |
3.66 |
βC18N5C19(22)+ ÓH4N3C18N5(77)+ ÓH6N5C19C21(79) |
| 34 |
349 |
337 |
|
338 |
1.28 |
1.10 |
βO35N34C7(26)+ βC20C19C21(58)+ βC7C8C37(65) |
| 35 |
339 |
328 |
|
316 |
0.48 |
3.63 |
βC20C19C21(58) |
| 36 |
247 |
239 |
|
238 |
0.63 |
1.81 |
βC16C20C25(42)+ ÓH32C29C26O2(83) |
Table 2: The selected fundamental vibrational assignments of CNPC.
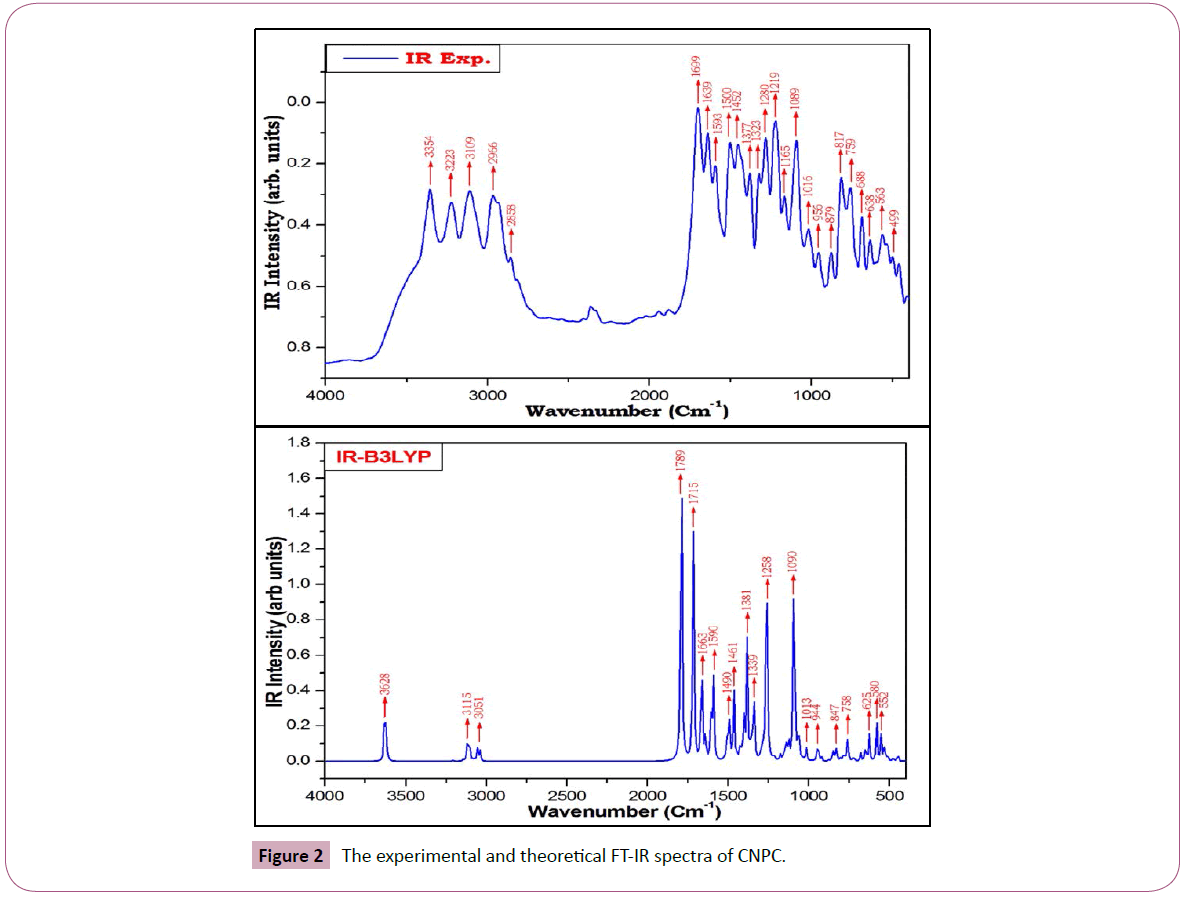
Figure 2: The experimental and theoretical FT-IR spectra of CNPC.
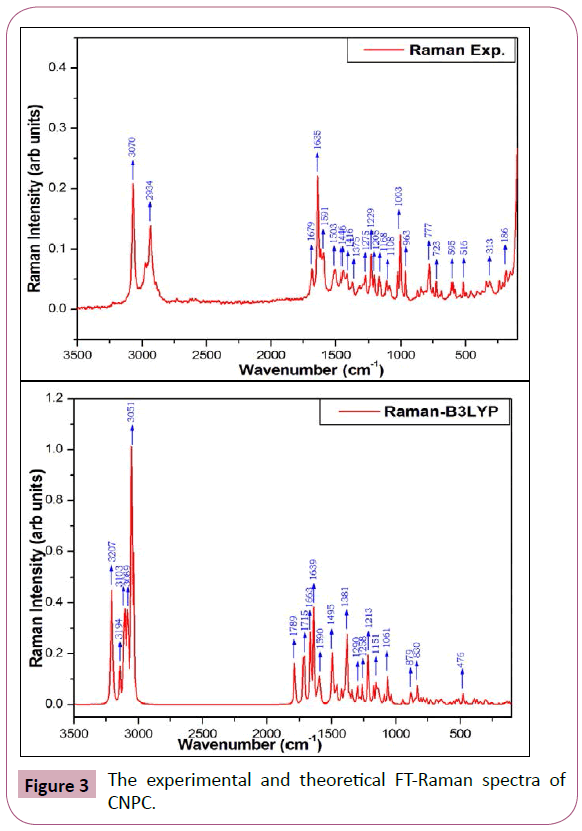
Figure 3: The experimental and theoretical FT-Raman spectra of CNPC.
C-Cl vibrations
The stretching and bending vibrations of C-Cl normally occur in the low wavenumber region 760-505 cm−1 [14]. Vibrational couplings are possible due to lowering of the molecular symmetry and the presence of heavy atom. The C-Cl stretching vibration of CNPC is observed as strong band at 688 cm−1 in FT-IR spectrum along with the vibration of βCCC bending vibration. The calculated wavenumber corresponds to the C-Cl stretching mode is 693 cm−1 with 68% of PED contribution. The chloro substituted aromatic compounds have a band of strong to medium intensity in the region 385-265 cm−1 due to C-Cl in-plane deformation [15]. The in-plane βCCCl is observed at 338 cm−1 in FT-Raman spectrum with medium intensity. The calculated in-plane and out-of-plane bending vibrations are computed at 297, 337 and 376 cm−1, respectively.
NO2 vibrations
The nitro stretching vibrations are the most characteristic bands in the spectra of nitro compounds, not only because of their spectral positions but also for their strong intensities. The nitro substituted aromatic compounds show asymmetric stretching mode in the region of 1600-1500 cm−1 and symmetric stretching mode in the region of 1385-1325 cm−1 [16]. In the present case, the asymmetric stretching vibrations of NO2 are observed at 1500 cm−1/FT-IR with medium intensity (27.45) and at 1502 cm−1/ FT-Raman spectrum. The calculated wavenumber at 1538 cm−1 is assigned for the asymmetric NO2 vibrations. The symmetric stretching vibration of NO2 is observed at 1323 cm−1 in FT-IR and calculated wavenumber computed at 1336 cm−1 with medium intensity. Aromatic nitro compounds show C−N stretching vibrations nearly ~870 cm−1 [16]. The theoretical band at 913 cm−1 is assigned for νC7−N34 vibration of the nitro group. The inplane deformation of NO2 is observed at 563 cm−1 in FT-IR and well agreed with the computed wavenumber at 562 cm−1.
N−H and C− N vibrations
In heterocyclic molecules, the N−H stretching vibrations are usually appearing in the region of 3500-3300 cm−1 [17]. The band observed at 3354 cm−1 in FT-IR spectrum is assigned for the N-H stretching vibration. The N3−H4 and N5−H6 stretching modes of dihydropyrimidinone have been calculated at 3501 and 3509 cm−1. The C-N stretching vibrations are normally occurring in the region 1400-1200 cm−1 [18]. The strong bands observed at 1229, 1205 cm−1 in FT-Raman and at 1219 cm−1 in FT-IR spectrum corresponds to the νC‒N stretching vibrations of the title molecule. These bands are in good agreement with the calculated wavenumber at 1235 and 1217 cm−1. The stretching wavenumber of C‒N associated with bending vibrations of βCNH and τCCCC vibrations. The bending vibrations of βCNH are calculated at 1446, 1442, 1414 and 1395 cm−1. Hence the corresponding experimental observation from FT-Raman shows wavenumber at 1446 and 1416 cm−1.
C=O and C−O vibrations
The carbonyl stretching vibrations are normally occurs in the region 1715-1600 cm−1, it is moderately active in Raman and intense in IR [19]. In the title molecule, the strong band observed at 1699 cm−1 in FT-IR with 100% IR intensity and 1679 cm−1 in FT-Raman is attributed to C=O stretching vibrations of dihydropyrimidine ring. The results of computation give the wavenumber of the corresponding mode at 1730 cm−1 has a contribution of 79% from C=O and minor contribution of 14% from C−N vibration. The C=O stretching vibration of ester group is observed at 1639 cm−1/ FT-IR and 1635 cm−1/FT-Raman. The corresponding computed wavenumber for C=O is 1659 cm−1 and the deviation is attributed to intermolecular C=O dipole-dipole interaction in the molecule. The bands responses to the ester C−O stretching vibrations are occur in the region of 1260-1000 cm−1 [20]. These vibrations are intense and partly due to an interaction with C−C vibration. The strong band observed at 1089 cm−1 in FT-IR and 1089 cm−1 in FT-Raman assigned to ester C−O stretching vibrations. It is well agreed with calculated wavenumber at 1086 cm−1.
C−H vibrations
The C−H stretching vibrations of aromatic ring absorb in its characteristic region 3100-3000 cm−1 [21]. The strong bands observed at 3109 cm−1 in FT-IR and 3070 cm−1 in FT-Raman confirms the C-H stretching vibrations of title molecule. The wavenumber computed at 3104, 3101 and 3089 cm−1 are well agreed with the experimental observations of C−H wavenumber. In the region below 3000 cm−1 are found aliphatic C−H stretching vibrations. The wavenumber corresponds to νC16−H17 is observed as strong band at 2966 cm−1 in IR spectrum and its calculated wavenumber shows 2976 cm−1 with 99% of PED contribution. In substituted phenyl rings, the C−H in-plane bending vibrational modes can be expected in the region 1300-1000 cm−1 [22]. In our study, the βCCH in-plane bending vibrations are computed at 1452, 1246, 1134 and 1113 cm−1. The strong band observed at 1452 cm−1 in FT-IR represents the C‒H in-plane bending vibration of the phenyl ring. The C‒H out of plane bending vibrations is expected to occur in the region 1000-675 cm−1 [23]. The C‒H out of plane bending vibration is observed as strong bands in both FT-IR and FT-Raman spectra at 956, 879, 817 and 963, 869 cm−1, respectively. These spectral wavenumber agreed with the calculated wavenumber at 953, 892, 827 and 819 cm−1.
Ring vibrations
The ring C‒C stretching vibrations occur in the region 1625- 1430 cm−1. In the phenyl ring, the six carbon atoms undergo coupled vibrations called skeletal vibration [24,25]. In present investigation, the ring C‒C stretching vibrations are observed as a strong band at 1593, 1503 cm−1 in FT-Raman and medium band at 1500 cm−1 in FT-IR are assigned for the ring vibrations. The theoretical wavenumber in the range 1585-1538 cm−1 represents the ring C‒C vibrations. The strong bands at 1168 and 1023 cm−1 in FT-Raman and weak band at 1165 cm−1 in FT-IR outcomes the phenyl ring breathing and trigonal bending vibration of the title molecule. These fundamental wavenumber computed at 1173 and 1026 cm−1 represents the ring breathing and trigonal bending vibrations of aromatic ring system.
CH3 and CH2 group vibrations
In methyl groups, the symmetric stretching vibrations are observed lower wavenumber when compared to the asymmetric stretching vibrations of C-H bonds. The CH3 symmetric vibrations are expected to occur in the range of 2950-2900 cm−1 and CH3 asymmetric stretching vibrations are expected in the range 3050- 2950 cm−1 [26,27]. The weak band observed at 2933 cm−1/FT-IR and strong band at 2934 cm−1/FT-Raman spectrum assigned for the symmetric stretching vibrations of the methyl group present in the carboxylate side chain. This mode has been calculated at 2936 cm−1. The symmetric C‒H vibrations of methyl and methylene group of CNPC calculated at 2955 and 2950 cm−1. The asymmetric vibrations of the methyl and methylene groups of title molecule computed in the region 3039-2987 cm−1. In many molecules, the symmetric deformations CH3 appears with an intensity varying from medium to strong and expected in the range 1380 ± 25 cm−1 [26]. This band has been observed at 1377 cm−1 in the FT-IR spectrum and 1375 cm−1 in FT-Raman spectrum. The rocking vibrations of methyl group usually observed in the region 1100 ± 95 cm−1 [28]. In present case, the medium band observed at 1016 cm−1 in FT-IR denotes the rocking vibration of methyl group attached to dihydropyrimidine rind and its corresponding computed wavenumber at 1019 cm−1. The rocking mode for the methyl group present in the carboxylate side chain shows weak band at 1157 cm−1 in FT-Raman spectrum and its calculated wavenumber at 1137 cm−1.
NBO Analysis
The molecular interactions occur in both the occupied and unoccupied molecular orbitals are examined by the Natural bond orbital analysis. Second order perturbation theory analysis of Fock matrix in NBO basis of CNPC is tabulated in Table 3. From the NBO results, two main orbital interactions are observed in the nitro group of the title molecule. The strong intra-molecular hyperconjugative interaction of LP(O35)→π*N34−O36 which increases the ED(0.5941) that weakens the respective anti-bonds leading to highest stabilization of 636.55 kJ/mol. From the Table 3, the stabilizing energy of n(LP3O35)→σ*(C7−N34), (N34−O36) and n(LP3O36)→σ*(C7−N34), (N34−O35) correspond to 52.17, 76.82 and 53.72, 80.92 kJ/mol, respectively. The strong intra-molecular hyperconjugative interaction of π(C−C)→π*(C−C) bonds of the phenyl ring increases ED at the six conjugated π-bonds. From the NBO analysis, the π-electron delocalization in phenyl ring revealed by the ED at the three conjugated π-bond (≈1.63- 1.67e) and π*(≈0.30-0.45e) resulting to the stabilization energies of ≈69.54-96.99 kJ/mol, respectively. The π-electron cloud movement can make the molecule highly polarized and causes internal charge transfer, which is responsible for the activity of the title molecule. The orbital overlap between n(Cl)→π*(C-C) bond orbital, which increases ED(0.4526) that weakens the respective anti-bonds (C7−C8=1.398Å) when compared to other C-C bonds of the phenyl ring leading to the stabilization of 62.38 kJ/mol. The most important interactions are n(LP2)O1→σ*O2−C25, n(LP2) O2→π*O1−C25, n(LP1)N3→π*C18−O33, n(LP1)N5→π*(C18−O33), (C19− C20) and n(LP2)O33→σ*N5−C18 provides stabilization energy of about 130.46, 185.94, 185.27, 160.92, 169.83 and 109.24 kJ/mol, respectively. The orbital occupancies of donor, acceptor and donor-acceptor interactions are visualized with the help of Chemcraft 1.8 software. The donor-acceptor interaction orbitals of CNPC were shown in Figure 4. These types of interactions are responsible for the pharmaceutical and biological activity of the title molecule.
| Type |
Donor |
ED/e |
Acceptor |
ED/e |
aE(2) kJ/mol |
bE(j)-E(i) a.u |
CF(i,j) a.u |
| π -π* |
C7-C8 |
1.6767 |
C9-C11 |
0.3025 |
69.54 |
0.32 |
0.065 |
| π -π* |
|
|
C13-C14 |
0.3247 |
79.66 |
0.32 |
0.069 |
| π -π* |
|
|
N34-O36 |
0.5941 |
58.16 |
0.18 |
0.047 |
| π -π* |
C9-C11 |
1.6405 |
C7-C8 |
0.4526 |
96.99 |
0.25 |
0.07 |
| π -π* |
|
|
C13-C14 |
0.3247 |
81.09 |
0.29 |
0.067 |
| π -σ* |
C13-C14 |
1.6381 |
N3-C16 |
0.0285 |
8.24 |
0.6 |
0.034 |
| π -π* |
|
|
C7-C8 |
0.4526 |
93.89 |
0.26 |
0.069 |
| π -π* |
|
|
C9-C11 |
0.3025 |
87.4 |
0.29 |
0.07 |
| π -π* |
C19-C20 |
1.8423 |
O1-C25 |
0.3047 |
97.11 |
0.29 |
0.076 |
| n -σ* |
LP(2) O1 |
1.8548 |
O2-C25 |
0.0958 |
130.46 |
0.62 |
0.126 |
| n -σ* |
|
|
C20-C25 |
0.0558 |
66.73 |
0.72 |
0.098 |
| n -σ* |
LP(1) O2 |
1.9637 |
O1-C25 |
0.0183 |
29.04 |
1.16 |
0.081 |
| n -π* |
LP(2) O2 |
1.8048 |
O1-C25 |
0.3047 |
185.94 |
0.33 |
0.112 |
| n -σ* |
LP(1) N3 |
1.7345 |
C13-C16 |
0.0456 |
30.17 |
0.65 |
0.065 |
| n -σ* |
|
|
C16-H17 |
0.0232 |
12.76 |
0.67 |
0.043 |
| n -σ* |
|
|
C18-O33 |
0.0439 |
10.46 |
0.83 |
0.043 |
| n -π* |
|
|
C18-O33 |
0.3284 |
185.27 |
0.33 |
0.11 |
| n -π* |
LP(1) N5 |
1.6803 |
C18-O33 |
0.3284 |
160.92 |
0.34 |
0.103 |
| n -π* |
|
|
C19-C20 |
0.2326 |
169.83 |
0.31 |
0.103 |
| n -σ* |
LP(2) O33 |
1.8465 |
N3-C18 |
0.0717 |
98.49 |
0.71 |
0.118 |
| n -σ* |
|
|
N5-C18 |
0.0860 |
109.24 |
0.65 |
0.118 |
| n -σ* |
LP(2) O35 |
1.8970 |
C7-N34 |
0.1027 |
52.17 |
0.55 |
0.074 |
| n -π* |
|
|
N34-O36 |
0.0629 |
76.82 |
0.72 |
0.104 |
| n -σ* |
LP(3) O35 |
1.4357 |
N34-O35 |
0.0683 |
25.52 |
0.7 |
0.067 |
| n -π* |
|
|
N34-O36 |
0.5941 |
636.55 |
0.16 |
0.141 |
| n -σ* |
LP(2) O36 |
1.8941 |
C7-N34 |
0.1027 |
53.72 |
0.55 |
0.075 |
| n -σ* |
|
|
N34-O35 |
0.0683 |
80.92 |
0.71 |
0.106 |
| n -π* |
LP(3) Cl37 |
1.9142 |
C7-C8 |
0.4526 |
62.38 |
0.31 |
0.067 |
a E(2) means energy of hyper conjugative interaction (stabilization energy).
b Energy difference between donor (i) and acceptor (j) NBO orbitals.
cF(i,j) is the fork matrix element between i and j NBO orbitals.
Table 3: The donor-acceptor interactions in NBO basis for CNPC.
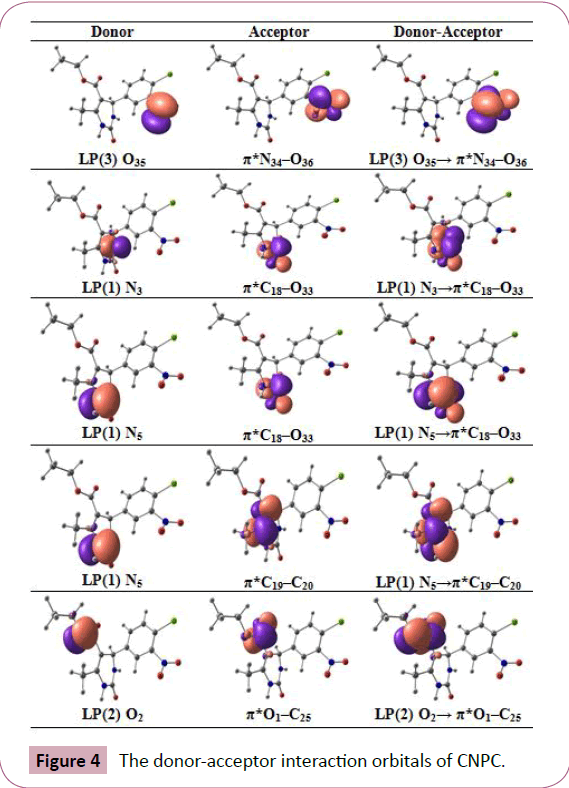
Figure 4: The donor-acceptor interaction orbitals of CNPC.
NLO Optics
Nonlinear optical studies is at the forefront of current research because of its prominence in providing the key functions of optical switching, optical modulation, optical logic and optical memory for the emerging technologies in areas such as signal processing, telecommunications and optical interconnections [29,30]. The molecular polarizability and hyperpolarizability are calculated about 4.71 × 10−30 esu and 2.61 × 10−30 esu, respectively.
The β0 value of the title compound is seven times greater than that of reference urea. Urea is one of the prototypical molecules used in the study of the NLO properties of molecular systems. Therefore, it was used frequently as a threshold value for comparative purposes [31]. The first hyperpolarizability values of similar pyrimidine derivatives are reported as, 1.19, 1.35 and 0.30 × 10−30 esu, respectively [32-34]. The first hyperpolarizability components of CNPC were listed in Table 4.
| Parameters |
B3LYP/6-31G(d,p) |
| Dipole moment ( μ ) Debye |
| μx |
2.5812 |
| μy |
-1.1982 |
| μz |
-0.1884 |
| μ |
2.8519 Debye |
| Polarizability ( α0 ) × 10-30esu |
| αxx |
246.60 |
| αxy |
8.27 |
| αyy |
239.80 |
| αxz |
1.66 |
| αyz |
21.69 |
| αzz |
177.97 |
| α |
4.7147 x10-30esu |
| Hyperpolarizability ( β0 ) × 10-30esu |
| βxxx |
-12.65 |
| βxxy |
11.97 |
| βxyy |
248.19 |
| βyyy |
-372.31 |
| βxxz |
-34.20 |
| βxyz |
-12.66 |
| βyyz |
37.17 |
| βxzz |
-97.30 |
| βyzz |
94.09 |
| βzzz |
-42.64 |
| β0 |
2.6143 × 10-30esu |
*Reference urea (μ=1.3732 Debye, β0=0.3728 × 10-30 esu)
Table 4: The first hyperpolarizability components of CNPC.
Molecular Electrostatic Potential
Molecular electrostatic potential is related to the electronic density of the molecule and is a very useful descriptor in understanding the charge sites as well as hydrogen bonding interactions [35,36]. The electrostatic potential V(r) are also well suited for analyzing processes based on the ‘‘recognition’’ of one molecule by another, as in drug-receptor, and enzyme substrate interactions, because it is through their potentials that the two species first ‘‘see’’ each other [37,38]. Being a real physical property V(r)s can be determined experimentally by diffraction or by computational methods [39]. The MEP mapped surface and 2D contour map of CNPC were shown in Figure 5. In order to predict the possible electrophilic and nucleophilic charge sites for the investigated molecule, MEP mapped surfaces are generated. The positive (blue) regions to nucleophilic reactivity and the negative (red and yellow) regions of MEP were related to electrophilic reactivity shown in Figure 5. The negative potentials are commonly observed in the region of electronegative atoms with lone pair electrons. In our study, the negative regions are localized in the oxygen atoms present in the nitro group and also with the oxygen atoms present in the two carbonyl groups, which are electrophilic nature. The positive potentials are localized over the hydrogen atoms bonded with the nitrogen atoms of dihydropyrimidinone ring, which are nucleophilic in nature.

Figure 5: The molecular electrostatic potential mapped surface of CNPC.
Energy Gap Analysis
Frontier molecular orbitals and their energies are very useful for the physicists and chemists. The analysis of the wave function indicates that the electron absorption corresponds to the transition from the ground to the first excited state and is mainly described by one-electron excitation from the highest occupied molecular orbital (HOMO) to the lowest unoccupied orbital (LUMO). The eigen values of HOMO/LUMO and their energy gap reflect the chemical activity of the molecule. In order to gain insight into the electronic structure of CNPC, it’s theoretical molecular orbital distributions were calculated with the Gaussian program at B3LYP/6-31G(d,p) level using the density functional theory. As shown in the Figure 6 the highest occupied molecular orbitals of CNPC were mostly dispersed on the 4-chloro-3-nitrophenyl moiety. In contrast, the LUMO were localized on the electron deficient pyrimidine ring together with the carboxylate side chain. The clear separation of the HOMO and LUMO suggested that the HOMO-LUMO excitation would shift the electron density distributions from the donor di-substituted phenyl moiety to the acceptor pyrimidine moiety leading to a polarized excited state. The contour map of title molecule describes the electron density mapped surface of the CNPC. It is more important, such separation of HOMO-LUMO can provide holes and electrontransporting channel, respectively.
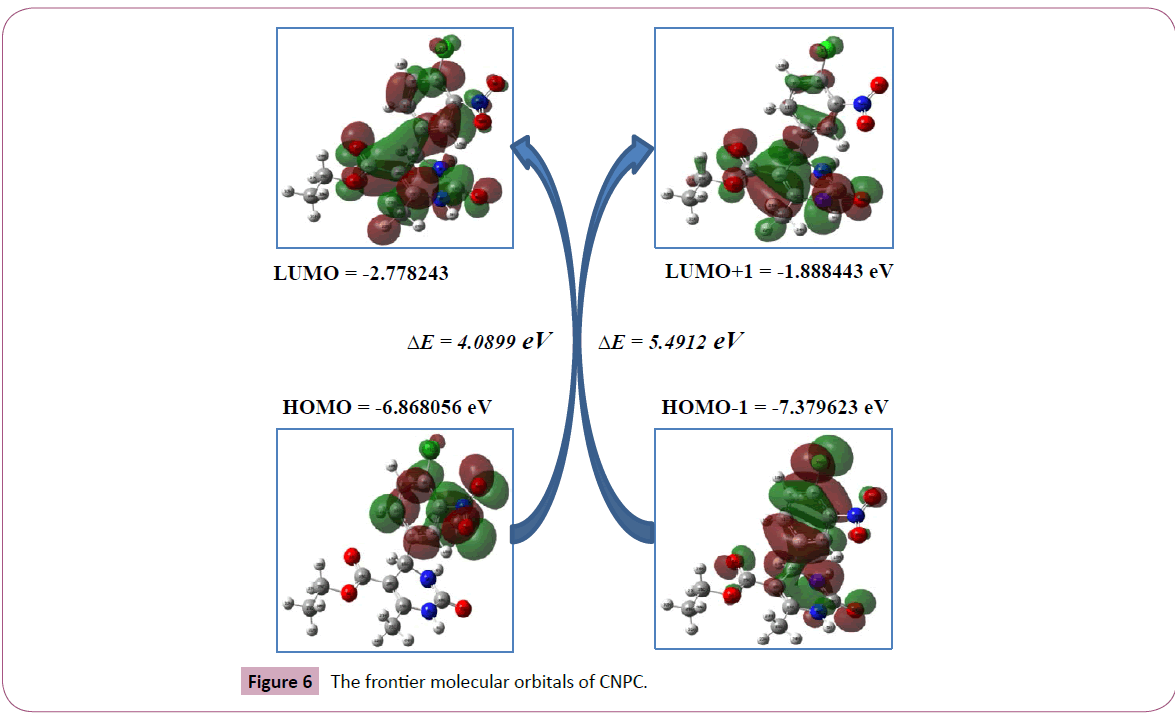
Figure 6: The frontier molecular orbitals of CNPC.
HOMO=-6.8682 eV
LUMO=-2.7783 eV
Energy gap ΔE=4.0899 Ev
NMR Analysis
Gauge independent atomic orbital method is used as a default method to calculate the NMR chemical shifts of the CNPC molecule. The recorded 1H-NMR and 13C-NMR spectra of the CNPC molecule are shown in Figure 7. Table 5, contains the assignments of experimental and theoretical chemical shift values of the title compound. The GIAO-NMR calculations were performed by B3LYP/6-31G(d,p) level of theory. Chemical shift of any ‘x’ proton (δX) is equal to the difference between isotropic magnetic shielding (IMS) of TMS and proton (x). It is defined by the equation: δX=IMSTMS–IMSX [40]. The theoretical NMR chemical shifts repositioned using proper scale factors. The 1H-NMR chemical shifts of CNPC (with respect to TMS) appeared in the range of 1.014-9.250 ppm experimentally and 0.862-7.456 ppm theoretically. The triplet and singlet observed at 1.037 and 2.225 ppm corresponds to the two methyl protons of the title molecule. These chemical shifts are in good accordance with the theoretical values at 1.064 and 2.373 ppm, respectively. The confirmation of the compound formation is observed by the singlet at 5.110 ppm in 1H-NMR chemical shift shows good agreement with the peak computed at 4.820 ppm. The quartet observed at the range of 3.354-3.988 ppm corresponds to the methylene group protons of the title molecule. Theoretical chemical shift for methylene group is computed in the range of 3.584-3.667 ppm, respectively. The experimental aromatic protons show nice accordance with the theoretical values.
| 1H-NMR |
|
| Atoms |
DFT |
Exp. |
Atoms |
DFT |
EXP. |
|
| C29 |
12.04 |
13.95 |
−C29H3 |
0.862-1.064 |
1.014-1.061 |
| C21 |
21.14 |
17.69 |
−C21H3 |
0.838-2.373 |
2.225 |
| C16 |
56.47 |
53.17 |
−C26H2 |
3.584-3.667 |
3.918-3.988 |
| C26 |
60.95 |
59.19 |
H17 |
4.820 |
5.11 |
| C14 |
125.14 |
117.30 |
ArC−H |
6.814-7.456 |
6.943-7.411 |
| C20 |
100.50 |
98.76 |
N−H |
3.886 |
7.781 |
| C8 |
134.99 |
119.81 |
N−H |
5.008 |
9.250 |
| C11 |
132.01 |
122.84 |
|
|
|
| C9 |
129.50 |
130.19 |
|
|
|
| C13 |
145.39 |
142.27 |
|
|
|
| C7 |
150.94 |
148.77 |
|
|
|
| C19 |
146.31 |
151.89 |
|
|
|
| C18 |
146.96 |
156.64 |
|
|
|
| C25 |
164.06 |
165.10 |
|
|
|
Table 5: The theoretical and experimental chemical shift values of CNPC.
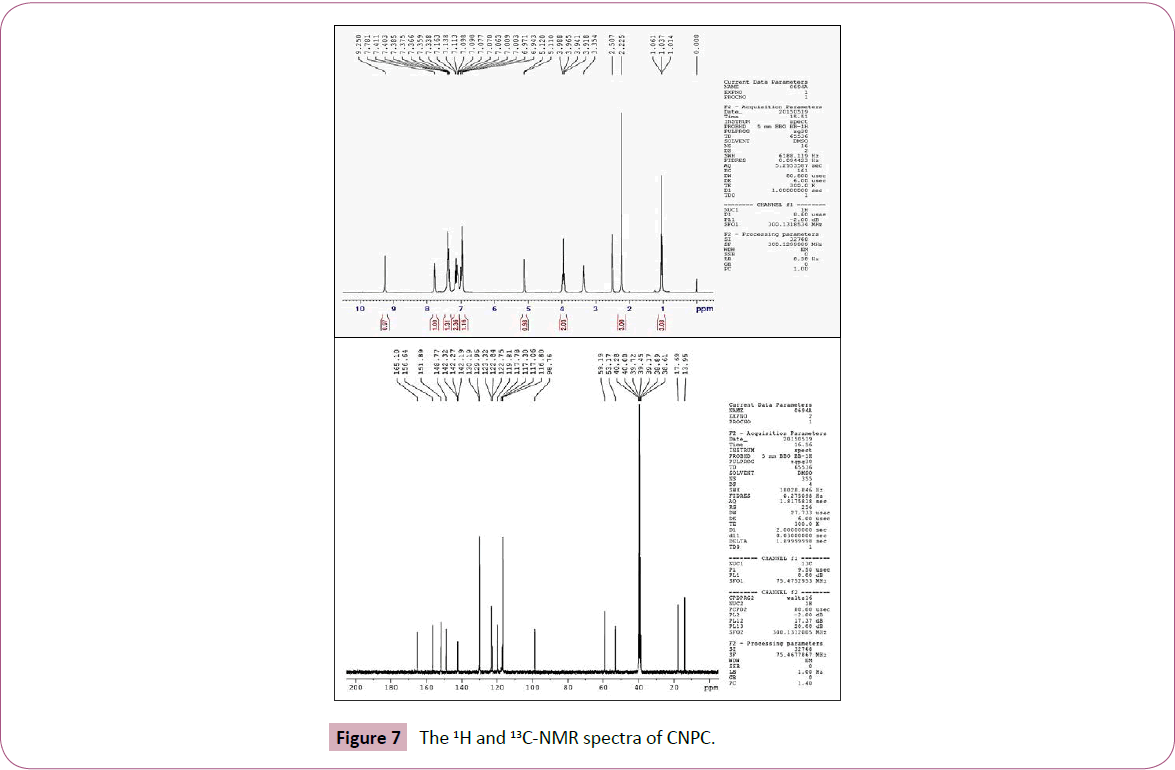
Figure 7: The 1H and 13C-NMR spectra of CNPC.
In 13C-NMR spectrum the fourteen signals confirms the fourteen carbons of the title molecule experimentally in the range of 13.95- 165.10 ppm and theoretically at 12.04-169.06 ppm, respectively. The signal observed at 53.17 ppm confirms the ring formation chiral carbon of the title molecule; its theoretical prediction coincides well with 56.47 ppm. The two signals 165.10 and 156.67 ppm at downfield region confirms the two carbonyl carbons (C25 and C18) of the CNPC molecule. These carbonyl carbon signals computed at 164.00 and 146.96 ppm. The peaks appeared at 13.95, 17.65 and 59.11 corresponds to the two methyl and one methylene carbon of the CNPC molecule. The calculated data of these carbons are 12.04, 21.14 and 60.95 ppm, respectively.
Conclusion
Nonlinear organic molecule ethyl-4-(4-chloro-3-nitrophenyl)-6- methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate has synthesized and characterized using spectral techniques. The stable conformer was identified with the relative energy of about -1542.44631 Hatree. Using potential energy distribution the vibrational modes of the recorded wavenumbers were assigned. The NBO analysis reveals the strong hyperconjugative interactions occur in the molecule leads to the stabilization of the molecular system. The small energy gap value 4.0899 eV is responsible for the nonlinear activity of the investigated molecule. The first hyperpolarizability calculated as 2.6143 x 10-30 esu, which is seven times greater than that of reference urea and their similar NLO molecules. Thus our title molecule is good candidate for the nonlinear optical studies. The molecular charge sites are identified by the molecular electrostatic potential mapped surface. The structural confirmation by 1H and 13C-NMR reports with computed chemical shifts shown very good agreement.
References
- Chemla DS, Zyss J (1987) In Nonlinear Optical Properties of Organic Molecules and Crystals, Academic Press: New York.
- Nalwa SH (2008) In Handbook of Organic Electronics and Photonics,American Scientific Publishers: Los Angeles.
- Mutter L, Koechlin M, Jazbinsek M, Gunter P (2007) Direct electron beam writing of channel waveguides in nonlinear optical organic crystals. Opt Expr 15: 16828-16838.
- Hwang EJ, Cheong CS, Lee H,Lee SW,Kim IT, Lee SH (2005) Synthesis and Photoluminescent Properties of Violet Emitting 5,6-Diphenylfuro[2,3-d]pyrimidine Derivatives. ChemInform 36: 47.
- Weng J, Mei Q, Fan Q, Ling Q, Tong B, Huang W (2013)Bipolar luminescent materials containing pyrimidine terminals: synthesis, photophysical properties and a theoretical study. RSC Adv3:21877-21887.
- Lin LY, Tsai CH, Wong KT, Huang TW, Wu CC, et al. (2011) Efficient organic DSSC sensitizers bearing an electron-deficient pyrimidine as an effective π-spacer J Mater Chem21:5950-5958.
- Achelle S, Rodríguez-López J, Cabon N, Guen FR (2015)Protonable pyrimidine derivative for white light emission RSC Adv5:107396-107399.
- Cui LS, Liu Y, Liu XY, Jiang ZQ, Liao LS (2015)Design and Synthesis of Pyrimidine-Based Iridium(III) Complexes with Horizontal Orientation for Orange and White Phosphorescent OLEDs ACS Appl Mater Interfaces 7:11007-11014.
- Gaussian 03 (2004) Revision C.02, Gaussian Inc., Wallingford, CT.
- Jamroz MH (2004) Vibrational Energy Distribution Analysis: VEDA4 program, Warsaw, Poland.
- Michalska D (2003) Raint Program, Wroclaw University of Technology.
- Michalska D, Wysokinski R (2005) The prediction of Raman spectra of platinum(II) anticancer drugs by density functional theory Chem. Phys. Lett., 403:211-217.
- Doddamani SB, Ramoji A, yenagi J, Tonamavar J (2007)The vibrational spectra, assignments and ab initio/DFT analysis for 3-chloro, 4-chloro and 5-chloro-2-methylphenyl isocyanates Spectrochim acta A 67:150-159.
- Socrates G (1981) IR characteristics group frequencies, John Wiley, New York.
- Roeges NPG (1994) A guide to complete interpretation of IR spectra of organic compounds, Wiley, New York.
- Barthes M, DeNunzio G, Ribet G (1996) Polarons or proton transfer in chains of peptide groups?Synth Met 76:337-340.
- Socrates G (2001)Infrared and Raman characteristic group frequencies – Tables and Charts, (3rd edn), Wiley, Chichester.
- Dhandapani A,Manivarman S, Subashchandrabose S, Saleem H (2014) Molecular structure and vibrational analysis on (E)-1-(3-methyl-2,6-diphenyl piperidin-4-ylidene) semicarbazide.J Mol Struct 1058: 41-50
- Dhandapani A, Manivarman S, Subashchandrabose S (2016) Synthesis, single crystal structure, Hirshfeld surface and theoretical investigations on pyrimidine derivative.Chem Phys Lett 655-656: 17-29.
- Lin-Vein D, Colthup NB, Fateley WG, Grasselli JG (1991) The Hand Book of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, San Diego.
- Varsanyi G (1969) Vibrational Spectra of Benzene Derivatives, Academic Press, New York.
- Silverstein RM, Clayton Bessseler G (1976)Spectroscopic Identification of organic compounds, Wiley, New York.
- Mohan J (2001) Organic Spectroscopy: Principles and Applications, (2nd edn), Narosa Publishing House, New Delhi.
- Bellamy LJ (1975) The Infrared Spectra of Complex Molecules, John Wiley & Sons, Inc, New York.
- Coates J (2000) in RA Meyers (Ed.), Interpretation of Infrared Spectra, A PracticalApproach, John Wiley and Sons Ltd, Chichester.
- Colthup NB, Daly LH, Wiberly SE (1990) Introduction to Infrared and Raman Spectroscopy, (3rd edn), Academic Press, Boston.
- Dollish FR, Fateley WG, Bentley FF (1997) Characteristic Raman Frequencies ofOrganic Compounds, John Wiley and Sons, New York.
- Nakano M, Fujita H, Takahata M, Yamaguchi K (2001)Theoretical Study on Second Hyperpolarizabilities of Phenylacetylene Dendrimer: Toward an Understanding of Structure−Property Relation in NLO Responses of Fractal Antenna Dendrimers.J Am Chem Soc 124: 9648-9655.
- Tanak H (2011) DFT computational modelling studies on 4-(2,3-Dihydroxybenzylideneamino)-3-methyl-1H-1,2,4-triazol-5(4H)-one.Comput Theoret Chem 967: 93-101.
- Ozdemir M, Sonmez M, Sen F, Dinçer M, Ozdemir N (2015) A novel one-pot synthesis of heterocyclic compound (4-benzoyl-5-phenyl-2-(pyridin-2-yl)-3,3a-dihydropyrazolo[1,5-c]pyrimidin-7(6H)-one): Structural (X-ray and DFT) and spectroscopic (FT-IR, NMR, UV-Vis and Mass) characterization Studies Spectrochim. Acta A137:1304-1314.
- Al-Abdullah ES, Mary YS, Panicker CY, El-Brollosy NR, El-Emam AA, et al. (2014) Theoretical investigations on the molecular structure, vibrational spectra, HOMO–LUMO analyses and NBO study of 1-[(Cyclopropylmethoxy)methyl]-5-ethyl-6-(4-methylbenzyl)-1,2,3,4-tetrahydropyrimidine-2,4-dione. Spectrochim acta A 133: 639-650.
- Panicker CY, Varghese HT, Eapen PE, Raju K, Ganguli S, et al. (2010) Spectroscopic investigations of 2-hydroxy-4-methyl pyrimidine hydrochloride.Int J Chem Sci 8:655-662.
- Pir H, Günay N, Tamer Ö, Avcı D, Atalay Y (2013)Theoretical investigation of 5-(2-Acetoxyethyl)-6-methylpyrimidin-2,4-dione: Conformational study, NBO and NLO analysis, molecular structure and NMR spectra.Spectrochim acta A112: 331-342.
- Scrocco E, Tomasi J (1978) Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials.Adv Quant Chem 11: 115-193.
- Luque FJ, Lopez JM, Orozco M (2000) Perspective on Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects.Theor Chem Acc 103: 343-345.
- Politzer P, Laurence PR, Jayasuriya K, McKinney J (1985) Molecular electrostatic potentials: an effective tool for the elucidation of biochemical phenomena Health Perspect. 61: 191-202.
- Scrocco E, Tomasi J (1973)The electrostatic molecular potential as a tool for the interpretation of molecular properties Topics in Current Chemistry. Springer 7:95-170.
- Tanak H, Alaman AA, Büyükgüngör O (2013) Combined experimental and DFT computational studies on (E)-1-(5-nitrothiophen-2-yl)-N-[4-(trifluoromethyl)phenyl] methanimine.J Mol Struct 1048: 41-50.
- Singh RN, Verma D, Kumar A, Baboo V (2012) Synthesis, molecular structure and spectral analysis of ethyl 4-[(3,5-dinitrobenzoyl)-hydrazonomethyl]-3,5-dimethyl-1H-pyrrole-2-carboxylate: A combined experimental and quantum chemical approach. Spectrochim Acta Part A 88:60-71.
- Konstantinov IA, Broadbelt LJ (2011)Regression Formulas for Density Functional Theory Calculated 1H and 13C NMR Chemical Shifts in Toluene-d8J. Phys Chem C 115:12364-12372.