Hannah Evangalin J1, Bharanidharan S2,* and Dhandapani A3
1Department of Physics, PRIST University, Chennai Campus, Mahabalipuram-603 102, Tamil Nadu, India
2Department of Physics, Bharath Institute of Higher Education and Research, Bharath University, Chennai-600 073, Tamil Nadu, India
3Department of Chemistry, CK College of Engineering and Technology, Cuddalore-607 003, Tamil Nadu, India
*Corresponding Author:
Bharanidharan S
Department of Physics, Bharath Institute of Higher Education and Research,
Bharath University, Chennai-600 073, Tamil Nadu, India.
Tel: +98 11 3368 7901
E-mail: bharani.dharan0@gmail.com
Received date: February 02, 2018; Accepted date: February 28, 2018; Published date: March 08, 2018
Citation: Evangalin JH, Bharanidharan S, Dhandapani A (2018) Molecular Spectroscopic Investigations of (E)-1-(4 Methylbenzylidene) Urea Using DFT Method. Arch Chem Res. Vol.2 No.2:18. doi:10.21767/2572-4657.100018
Copyright: © 2018 Evangalin JH, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords
DFT Study; FT-IR; FT-Raman; PED; NMR; NLO
Introduction
Schiff base compounds have been derived from aromatic amines and aromatic aldehydes. Schiff-base compounds exhibit a wide range of biological activities [1], anti-HIV [2], anti-tumor activities [3] and the transition metal complexes [4,5]. Very often it undergoes a variety of chemical reactions, being as a key precursor for new compounds exhibiting diverse molecular structures and properties [6,7]. It is worth mentioning that the salicylaldehyde moiety appears in many compounds exhibiting various biological activity, including reactants used in the design of compounds exhibiting anti-viral activity [8], as well as in reactions resulting in new compounds with anti-cancer [9,10] or anti-microbial activity [11]. It is also present during the synthesis of new products called “aspirin-like molecules” exhibiting antiinflammatory activity [12].
The computational techniques are recently very useful in exploring nonlinear properties theoretically in useful manner. The simultaneous researches in the field of nonlinear materials have influenced the development of efficiencies of organic molecules as NLO material, since organic NLO materials often employ materials and device structures that have direct application to light multiplication.
In our present investigation we have focused on a simple organic molecule (E)-1-(4-methylbenzylidene) urea. We planned to investigate the nonlinear property of that molecule theoretically using first hyperpolarizability calculation. In addition to that, the vibrational spectral studies, NLO activity, NBO analysis, MEP surface and energy gap of the molecule are studied and discussed in detail.
Experimental Details
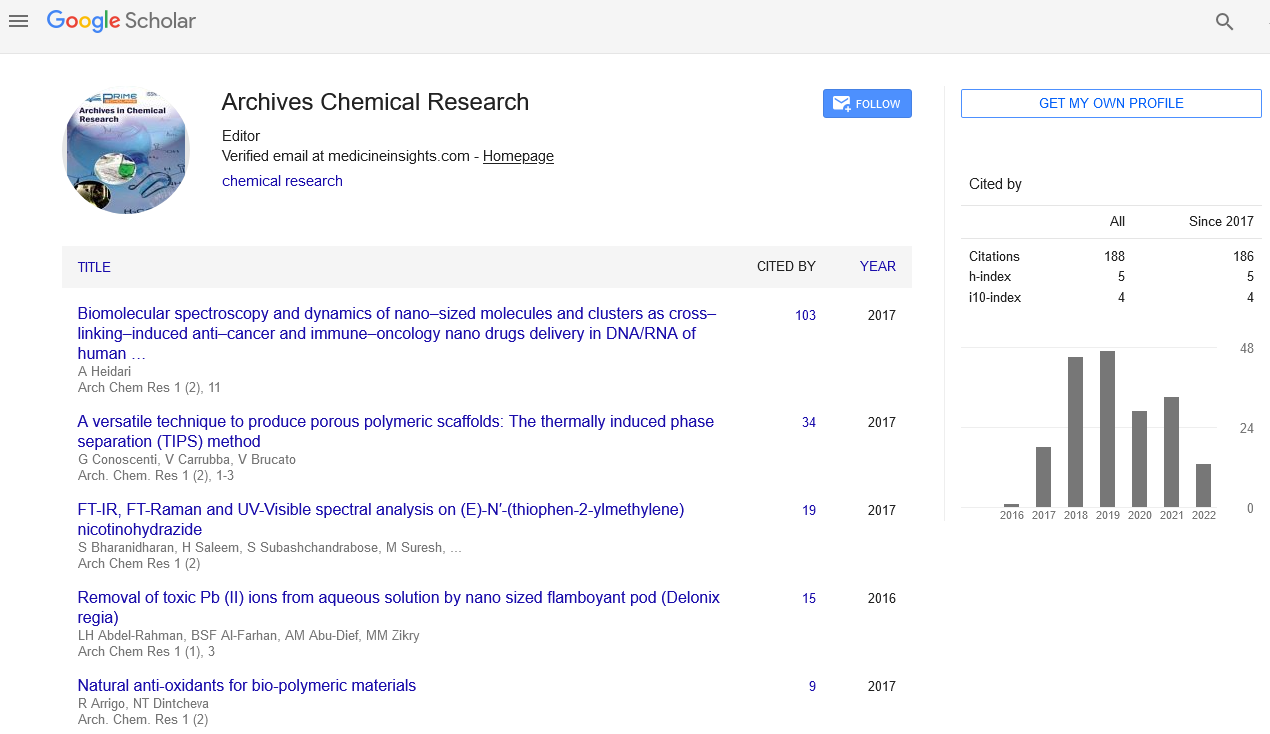
Synthesis of (E)-1-(4-Methylbenzylidene)urea
Equimolar amount of 4-methylbenzaldehyde and urea were dissolved in 30 ml of absolute ethanol. The mixture was shaken to make homogenous solution. Few drops of catalyst acetic acid were added to increase the rate of reaction. The content was refluxed at 90°C for 3 hours. The completion of the reaction was monitored by thin layer chromatography. After the reaction was completed, the content was cooled the mixture was poured into water. The solid product obtained was filtered and purified using absolute ethanol
Spectral measurements
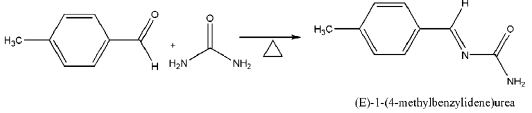
FT-IR, FT-Raman and NMR Spectrum: The FT-IR spectrum of the synthesized EMBU molecule was measured in the Mid- IR (4000–400 cm-1) region at the spectral resolution of 4 cm-1 using on SHIMADZU FT-IR affinity Spectrophotometer (KBr pellet technique) in Faculty of Marine Biology, Annamalai University, Parangipettai. The FT-Raman spectrum was recorded on BRUKER: RFS27 spectrometer operating at laser 100mW in the spectral range of 4000–50 cm-1. FT-Raman spectral measurements were carried out from SAIF, IIT, Chennai. NMR spectral studies were carried out using Bruker 400 MHz spectrometer, using TMS as an internal standard and DMSO-d6 as solvent and recorded at Annamalai University, Annamalainagar, Chidambaram. The 1H and 13C-NMR spectrum were shown in Figure 1.
Figure 1: The 1H and 13C-NMR spectra of EMBU.
Computational Details
The quantum chemical calculations were performed at DFT method with B3LYP/6-31G(d,p) basis set using Gaussian 03W [13] program package, invoking gradient geometry optimization [13,14]. The optimized structural parameters were used in the vibrational frequency calculations at the DFT level to characterize all the stationary points as minima. The vibrational modes were assigned on the basis of PED analysis using VEDA4 program package [15]. NBO analysis was done by using NBO 3.1 program.
The Raman activity was calculated by using Gaussian 03W package and the activity was transformed into Raman intensity using Raint program [16] by the expression:
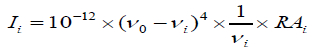 (1)
(1)
Where Ii is the Raman intensity, RAi is the Raman scattering activities, νi is the wavenumber of the normal modes and ν0 denotes the wavenumber of the excitation laser [17].
Results and Discussion
Molecular geometry
The Optimized structure of EMBU molecule is shown in Figure 2 with atom numbering scheme. The bond parameters such as bond lengths, bond angles and dihedral angles values are calculated by DFT/B3LYP/6-31G(d,p) basis set and are listed in Table 1. In this case, the bond length of C-C and C-H are calculated around 1.40 and 1.08 Å, respectively. The bond lengths of related molecule was observed at C-C and C-H values are 1.38, 1.09 Å, respectively [18]. The carbonyl group bond C13=O18 is appeared as π bond character; its bond length value is calculated about 1.2182 Å. Similarly, the π bond character of C=O is observed at 1.212 Å [19], which nearly coincides with calculated value. The σ and π bond characters of N12-C13 and C11=N12 are calculated as 1.4341 and 1.281Å are positively deviated with related molecule data (1.348 Å and 1.273 Å) data [19].
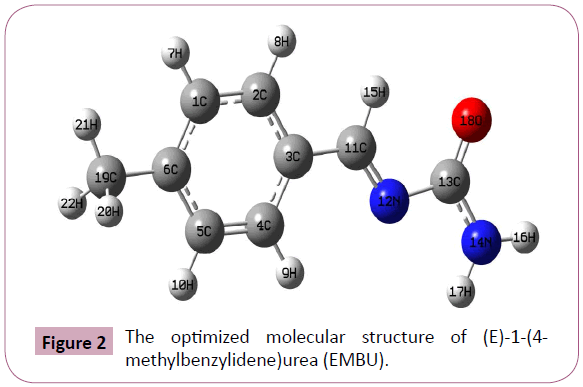
Figure 2: The optimized molecular structure of (E)-1-(4- methylbenzylidene)urea (EMBU).
| Bond Lengths (Å) |
B3LYP/
6-31G(d,p) |
Bond Angles (°) |
B3LYP/
6-31G(d,p) |
Dihedral Angles (°) |
B3LYP/
6-31G(d,p) |
| R(1,2) |
1.3921 |
A(2,1,6) |
120.8487 |
D(6,1,2,3) |
0.0032 |
| R(1,6) |
1.3973 |
A(2,1,7) |
119.6136 |
D(6,1,2,8) |
179.9973 |
| R(1,7) |
1.0848 |
A(6,1,7) |
119.5377 |
D(7,1,2,3) |
-179.994 |
| R(2,3) |
1.4003 |
A(1,2,3) |
120.6796 |
D(7,1,2,8) |
-0.0001 |
| R(2,8) |
1.0853 |
A(1,2,8) |
119.8559 |
D(2,1,6,5) |
-0.0096 |
| R(3,4) |
1.4055 |
A(3,2,8) |
119.4645 |
D(2,1,6,19) |
179.957 |
| R(3,11) |
1.4605 |
A(2,3,4) |
118.6712 |
D(7,1,6,5) |
179.9879 |
| R(4,5) |
1.3847 |
A(2,3,11) |
119.2354 |
D(7,1,6,19) |
-0.0456 |
| R(4,9) |
1.083 |
A(4,3,11) |
122.0934 |
D(1,2,3,4) |
0.003 |
| R(5,6) |
1.4056 |
A(3,4,5) |
120.3294 |
D(1,2,3,11) |
179.9981 |
| R(5,10) |
1.0856 |
A(3,4,9) |
118.7107 |
D(8,2,3,4) |
-179.991 |
| R(6,19) |
1.5076 |
A(5,4,9) |
120.9599 |
D(8,2,3,11) |
0.004 |
| R(11,12) |
1.281 |
A(4,5,6) |
121.223 |
D(2,3,4,5) |
-0.0026 |
| R(11,15) |
1.0958 |
A(4,5,10) |
119.5125 |
D(2,3,4,9) |
179.9914 |
| R(12,13) |
1.4341 |
A(6,5,10) |
119.2645 |
D(11,3,4,5) |
-179.998 |
| R(13,14) |
1.3596 |
A(1,6,5) |
118.2481 |
D(11,3,4,9) |
-0.0036 |
| R(13,18) |
1.2182 |
A(1,6,19) |
121.3126 |
D(2,3,11,12) |
-179.999 |
| R(14,16) |
1.0061 |
A(5,6,19) |
120.4392 |
D(2,3,11,15) |
0.0028 |
| R(14,17) |
1.0058 |
A(3,11,12) |
123.1686 |
D(4,3,11,12) |
-0.0038 |
| R(19,20) |
1.0945 |
A(3,11,15) |
116.9094 |
D(4,3,11,15) |
179.9978 |
| R(19,21) |
1.0914 |
A(12,11,15) |
119.922 |
D(3,4,5,6) |
-0.004 |
| R(19,22) |
1.0944 |
A(11,12,13) |
115.0758 |
D(3,4,5,10) |
179.9926 |
| |
|
A(12,13,14) |
110.1617 |
D(9,4,5,6) |
-179.998 |
| |
|
A(12,13,18) |
126.0669 |
D(9,4,5,10) |
-0.0012 |
| |
|
A(14,13,18) |
123.7714 |
D(4,5,6,1) |
0.01 |
| |
|
A(13,14,16) |
119.3699 |
D(4,5,6,19) |
-179.957 |
| |
|
A(13,14,17) |
119.9243 |
D(10,5,6,1) |
-179.987 |
| |
|
A(16,14,17) |
120.7058 |
D(10,5,6,19) |
0.0466 |
| |
|
A(6,19,20) |
111.0369 |
D(1,6,19,20) |
-119.878 |
| |
|
A(6,19,21) |
111.4884 |
D(1,6,19,21) |
0.5522 |
| |
|
A(6,19,22) |
111.0578 |
D(1,6,19,22) |
121.0241 |
| |
|
A(20,19,21) |
107.9612 |
D(5,6,19,20) |
60.0881 |
| |
|
A(20,19,22) |
107.1337 |
D(5,6,19,21) |
-179.482 |
| |
|
A(21,19,22) |
107.983 |
D(5,6,19,22) |
-59.0101 |
| |
|
|
|
D(3,11,12,13) |
-179.998 |
| |
|
|
|
D(15,11,12,13) |
0.0003 |
| |
|
|
|
D(11,12,13,14) |
-179.996 |
| |
|
|
|
D(11,12,13,18) |
0.0049 |
| |
|
|
|
D(12,13,14,16) |
179.9973 |
| |
|
|
|
D(18,13,14,17) |
-179.998 |
Table 1: The optimized bond parameters of EMBU.
The bond angles of N12-C13-O18 and N14-C13=O18 is calculated about 126.06 and 123.77°, respectively. Similarly, the reported molecule calculated the bond angle value of N-C=O is 123.36°, which nearly matches with calculated values [18]. These results fairly explore that, during the course of rotation of the bond to the entire molecule gets disturbed and the properties of the molecule also changes. From the literature [20] and our theoretical investigation, the optimized structure is more stable.
Vibrational assignments
The EMBU molecule belongs to C1 point group symmetry. It consists of 22 atoms and 60 normal modes of vibrations are distributed among the symmetry species as;
Γ3N-6 = 41′ (in-plane) + 19′′ (Out-of-plane)
All these modes are found to be are active both in the IR and Raman absorption. The harmonic wavenumbers are calculated using B3LYP/6-31G(d,p) basis set and compared with recorded vibrational frequencies of FT-IR and FT-Raman spectra, respectively. Some discrepancies could be identified in between harmonic and observed frequencies, which are scaled down by proper scale factor [21,22]. The vibrational assignments are given in Table 2. The combined recorded and theoretical spectra are shown in Figures 3 and 4.
| Mode No |
Theoretical |
Experimental |
IR Intb |
Raman Intc |
Vibrational Assignments PED≥(10%)d |
| Un Scaled |
Scaleda |
IR |
Raman |
| 1 |
3745 |
3603 |
3606 |
|
16.8 |
0.9 |
νN14H16(100)+νN14H17(100) |
| 2 |
3608 |
3471 |
3460 |
|
20.4 |
2.5 |
νN14H16(100)+νN14H17(100) |
| 3 |
3199 |
3077 |
|
|
0.7 |
1.4 |
νC4H9(95) |
| 4 |
3177 |
3056 |
3061 |
|
3.8 |
3.2 |
νC1H7(97)+νC2H8(95) |
| 5 |
3160 |
3040 |
|
|
1.6 |
1.8 |
νC1H7(97)+νC2H8(95) |
| 6 |
3160 |
3040 |
|
|
4.3 |
1.5 |
νC5H10(89) |
| 7 |
3107 |
2989 |
2987 |
|
4.6 |
1.6 |
νC19H21(99) |
| 8 |
3074 |
2957 |
|
|
3.5 |
2.7 |
νC19H20(90)+νC19H22(90) |
| 9 |
3052 |
2936 |
2929 |
|
2.8 |
1.1 |
νC11H15(100) |
| 10 |
3025 |
2910 |
|
|
6.8 |
9.8 |
νC19H20(90)+νC19H21(99)+νC19H22(90) |
| 11 |
1762 |
1695 |
1672 |
1648 |
111.6 |
1.5 |
νO18C13(88) |
| 12 |
1671 |
1607 |
|
|
52.2 |
33.9 |
νN12C11(66) |
| 13 |
1646 |
1583 |
1598 |
|
45.4 |
100 |
νC4C5(53)+νC2C1(62)+νC2C3(48) |
| 14 |
1602 |
1541 |
|
1539 |
0.4 |
0.9 |
νC4C3(47)+νC6C5(79)+βH16N14H17(83)+βC2C1C6(57) |
| 15 |
1595 |
1534 |
1506 |
|
117.4 |
27.1 |
βH16N14H17(83) |
| 16 |
1543 |
1484 |
|
1468 |
0.9 |
3.5 |
βH7C1C2(54)+βH9C4C5(66)+βH10C5C4(75)+βC4C3C2(65) |
| 17 |
1494 |
1438 |
1435 |
|
4.5 |
4.9 |
βH21C19C6(76)+βH20C19H22(84) |
| 18 |
1486 |
1429 |
|
|
2.1 |
1.4 |
βH20C19C6(85)+?C19H20C6H21(91) |
| 19 |
1440 |
1385 |
|
|
4.5 |
2.9 |
νC4C5(53)+νC2C1(62) |
| 20 |
1415 |
1361 |
1354 |
|
0.4 |
4.4 |
βH20C19H22(84)+?C19H21H22H20(80) |
| 21 |
1400 |
1347 |
|
|
18.4 |
2.8 |
νN12C11(66)+βH15C11N12(82) |
| 22 |
1341 |
1290 |
1298 |
|
28.1 |
2.4 |
νN14C13(55)+βH9C4C5(66)+βH10C5C4(75) |
| 23 |
1338 |
1287 |
|
|
0.7227 |
1.3 |
νC2C3(48)+νC6C5(79) |
| 24 |
1325 |
1275 |
|
|
100 |
15 |
νN14C13(55)+βH16N14C13(56)+βH15C11N12(82) |
| 25 |
1246 |
1199 |
|
|
31.2 |
30 |
νC11C3(50)+βH7C1C2(54)+βH10C5C4(75)+βH15C11N12(82) |
| 26 |
1229 |
1182 |
|
1174 |
3.9 |
9.6 |
νC2C1(62)+βC1C6C5(51)+νC19C6(64) |
| 27 |
1198 |
1152 |
|
|
19.8 |
22.5 |
βH7C1C2(54)+βH8C2C1(50)+βH9C4C5(66)+βH10C5C4(75) |
| 28 |
1138 |
1095 |
|
|
1.7 |
1.1 |
νC4C5(53)+νC2C1(62)+βH7C1C2(54)+βH8C2C1(50)+βH9C4C5(66) |
| 29 |
1098 |
1056 |
|
|
1.1 |
8.5 |
νO18C13(88)+νN14C13(55)+βH16N14C13(56) |
| 30 |
1061 |
1021 |
|
|
1.5 |
0 |
βH20C19C6(85)+?C19H20C6H21(91) |
| 31 |
1053 |
1013 |
1002 |
1009 |
0.9 |
0.9 |
τH15C11N12C13(82) |
| 32 |
1035 |
996 |
|
|
2.3 |
0.3 |
βC1C6C5(51)+βC2C1C6(57)+βC6C5C4(57) |
| 33 |
1008 |
969 |
|
|
4.1 |
0.6 |
νC6C5(79)+βH21C19C6(76) |
| 34 |
999 |
962 |
950 |
|
0 |
0 |
?C4C3C5H9(81)+τH10C5C4C3(85) |
| 35 |
971 |
934 |
|
|
0.1 |
0 |
τH7C1C2H8(77) |
| 36 |
935 |
899 |
|
|
31 |
4.6 |
νN12C13(51)+βN12C11C3(75) |
| 37 |
874 |
840 |
867 |
|
0 |
6.8 |
νC2C3(48)+νC4C3(47)+νC11C3(50) |
| 38 |
856 |
824 |
829 |
|
0.9 |
0 |
τH7C1C2C3(82)+?C4C3C5H9(81)+τH10C5C4C3(85) |
| 39 |
838 |
806 |
|
|
11 |
0 |
τH7C1C2C3(82)+τH10C5C4C3(85) |
| 40 |
786 |
756 |
|
|
0.2 |
6.6 |
νC19C6(64)+βC4C3C2(65) |
| 41 |
780 |
750 |
746 |
|
0.1 |
0 |
τC3C11N12C13(68)+?O18N12N14C13(70) |
| 42 |
723 |
695 |
|
|
1.3 |
0.2 |
τC1C6C2C3(82)+τC3C2C4C5(83)+τC1C6C4C5(77) |
| 43 |
657 |
632 |
|
|
1.9 |
1.6 |
νC6C5(79)+βN12C13O18(65)+βC6C5C4(57) |
| 44 |
647 |
623 |
613 |
|
3.6 |
2.9 |
βC1C6C5(51)+βN12C13O18(65) |
| 45 |
608 |
585 |
|
546 |
0.2 |
0 |
?H16N14C13N12(93) |
| 46 |
563 |
542 |
|
|
1 |
0.8 |
βN12C11C3(75)+βN14C13N12(80)+βC2C3C11(68) |
| 47 |
512 |
492 |
|
|
8.7 |
0.1 |
τC3C2C4C5(83)+τC1C6C4C5(77) |
| 48 |
509 |
489 |
476 |
|
3.4 |
0.2 |
βC1C6C5(51)+βN14C13N12(80) |
| 49 |
422 |
406 |
|
|
0.1 |
0.1 |
τC1C6C2C3(82)+τC1C6C4C5(77) |
| 50 |
371 |
357 |
|
|
1.6 |
0.8 |
βN14C13N12(80)+βC2C3C11(68)+βC5C6C19(75) |
| 51 |
352 |
339 |
|
|
0.9 |
1.4 |
τC3C2C4C5(83)+τC4C5C6C19(68)+τC1C2C3C11(79) |
| 52 |
298 |
287 |
|
|
0.5 |
1.8 |
βN14C13N12(80)+βC2C3C11(68)+βC5C6C19(75) |
| 53 |
240 |
231 |
|
|
1.2 |
3.2 |
νC11C3(50)+βN12C13O18(65)+βC4C3C2(65)+βC13N12C11(61) |
| 54 |
230 |
221 |
|
|
61 |
0.5 |
?N14H16C13H17(89) |
| 55 |
189 |
182 |
|
|
0 |
0.8 |
τC4C3C11N12(77)+τC3C2C4C5(83)+τC4C5C6C19(68) |
| 56 |
151 |
146 |
|
132 |
0.9 |
0.3 |
τC4C3C11N12(77)+τN14C13N12C11(95)+τC3C11N12C13(68) |
| 57 |
94 |
91 |
|
101 |
0.3 |
2.6 |
βN12C11C3(75)+βC2C3C11(68)+βC13N12C11(61) |
| 58 |
63 |
60 |
|
63 |
0 |
2.2 |
τC3C11N12C13(68)+τC1C2C3C11(79) |
| 59 |
32 |
31 |
|
|
2.3 |
1.5 |
τC4C3C11N12(77)+τN14C13N12C11(95) |
| 60 |
15 |
15 |
|
|
0 |
22.8 |
τH22C19C6C5(91) |
ν: Stretching, β: in-plane-bending, Γ: out-of-plane bending, τ: Torsion,
aScaling factor: 0.9620,
bRelative IR absorption intensities normalized with highest peak absorption equal to 100,
cRelative Raman intensities normalized to 100,
dPotential energy distribution calculated at B3LYP/6-31G(d,p) level.
Table 2: The fundamental vibrational assignments of EMBU.
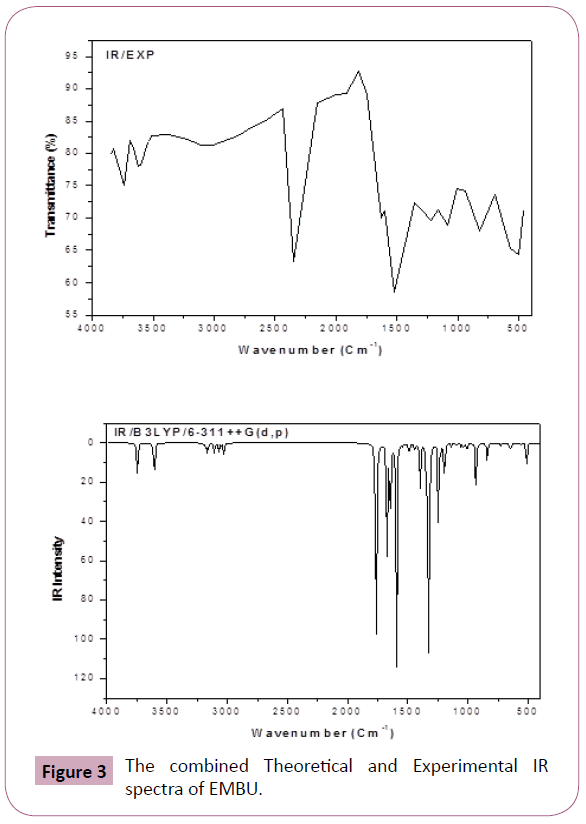
Figure 3: The combined Theoretical and Experimental IR spectra of EMBU.
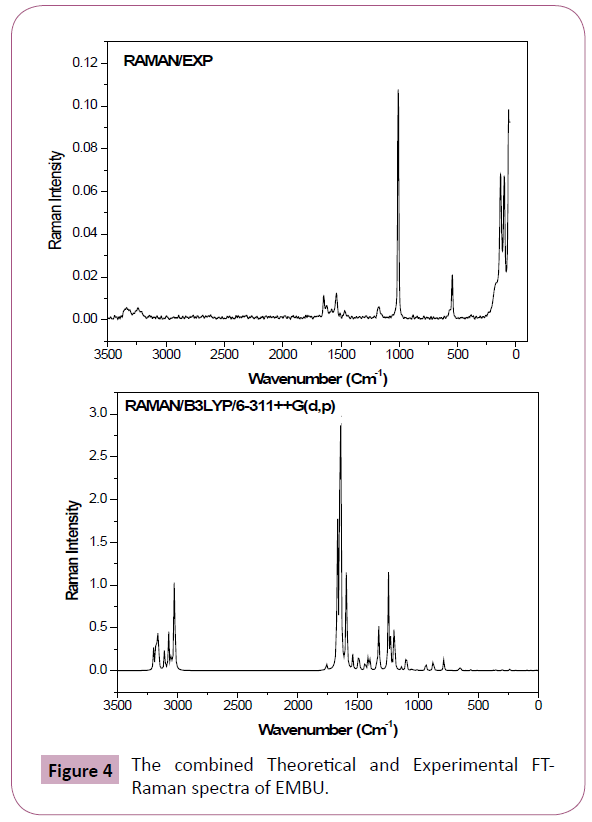
Figure 4: The combined Theoretical and Experimental FTRaman spectra of EMBU.
C=O Vibrations: The C=O stretching vibrations has been most extensively studied by IR and Raman spectroscopy. This multiply bonded group is highly polar and therefore gives rise to an intense IR absorption band, because of the different electro negativities are distributed between the two atoms [23]. The lone pair oxygen atom also determines the nature of the carbonyl group. In this study, the weak bands observed at 1672 and 1648 cm-1 in FTIR and FT-Raman spectra are assigned to C=O stretching vibration and its corresponding harmonic value lies at 1692 cm-1 (mode no: 11). This vibrational assignment is further supported by PED 88%.
C-C Vibrations: Generally, the phenyl ring carbon-carbon (C-C) stretching vibrations are observed in the region of 1625-1590, 1590-1575, 1540-1470, 1465-1430 and 1380-1280 cm-1 by Varsanyi (1974) [24]. In the present study, the FT-Raman bands observed at 1539 and FTIR band observed at 1598 cm-1 and are assigned to C-C stretching vibrations and their corresponding harmonic values appeared in the range of 1583 and 1541 cm-1 (mode nos: 13 and 14). These assignments are good agreement with literature [25].
The C-C in-plane bending vibrations observed at 1174 cm-1 in Raman and 613 cm-1 in IR spectrum and its corresponding calculated value appeared at 1182 and 623 cm-1 (mode no: 26 and 44). The ?C4C3C5H9 mode predicted at 962 cm-1 (mode no: 34) is in agreement with IR band observed at 950 cm-1 with the help of PED 81%.
C-H Vibrations: In General, the CH stretching modes are expected to occur in the range 2900-3200 cm−1 [26,27]. In our study, C-H vibrations are observed at 3061, 2987 and 2929 cm-1 in FTIR spectrum. The corresponding harmonic frequencies lies at 3056, 2989 and 2936 cm-1 (mode nos: 4, 7 and 9) are belong to the same mode. These results were good agreement with experimental value with PED >95%. In benzene like molecule the C-H inplane bending vibrations are appeared in the region 1000-1300 cm−1 and are usually weak intense. In our study, the frequency of the βCH vibrations are calculated in the region of 1438-1290 cm-1 (mode nos: 16 and 22) for this molecule. These modes are observed in the FTIR: 1298 cm-1/FT-Raman: 1468 cm-1 spectra with weak intensity. The harmonic frequencies in the range 962 cm-1 (mode nos: 34) and FTIR band observed at 950 cm-1 assigned to ΓCH mode. These assignments are find support from PED.
N-H Vibrations: In general, the N-H stretching vibrations observed in the region 3400-3200 cm-1 [28]. In the present case, the N-H band assigned at 3460 cm-1 (mode no: 2). This assignment is further justified on the basis of their calculated PED value (100 %). The calculated wavenumber for βN-H (1534 cm-1/ mode no: 15) and ΓN-H (585 cm-1/ mode no: 45) modes well reproduced the experimental ones in FT-IR (1506 cm-1) and FT-Raman (546 cm-1) spectra with PED values (83% and 93%), respectively.
C-N Vibrations: The assignment of C=N stretching frequency is a rather difficult task since there are problems in identifying these frequencies from other vibrations. The ring C–N vibrations are appeared in the region 1650–1550 cm−1 [29]. The bands obtained at 1598 cm-1 in FT-IR and at 1539 cm-1 in FT-Raman spectra have been assigned to C=N stretching vibrations of title molecule. The corresponding theoretical wavenumber lies at 1583 and 1541 cm-1, which is comparable to experimental wavenumber with PED contribution ≥ 45%. Silverstein et al., [30] assigned C–N stretching vibrations occurred in the region 1382–1266 cm-1 for the aromatic amines. The C-N stretching vibrations of title molecule were obtained at 1298 cm-1 in FT-IR spectrum. The band calculated at 1290 cm-1 from DFT is assigned to C-N stretching vibration of the present molecule.
NLO property
In this study, the electronic dipole moment, molecular polarizability, anisotropy of polarizability and molecular first hyperpolarizability of EMBU were calculated at DFT/B3LYP/6- 31G(d,p) basis set and are presented in Table 3. It is well-known that the higher values of dipole moment, molecular polarizability, and hyperpolarizability which enhances the NLO property. Urea is one of the prototypical molecule used in the study of the NLO property of molecular systems, and thus, it was used frequently as a threshold value for comparative purposes. The first hyperpolarizability value of EMBU was calculated as 10.595 × 10−30 esu. According to these result, the β0 value of present molecule is twenty-eight times larger than the magnitude of urea, which implies that the title molecule might become a kind of good NLO material.
| Parameters |
B3LYP/6-31G(d,p) |
| Dipole moment ( μ ) Debye |
| μx |
-1.1362 |
| μy |
-0.8895 |
| μz |
0.0003 |
| μ |
1.4429 Debye |
| Polarizability ( α0 )x10-30esu |
| αxx |
214.95 |
| αxy |
-4.99 |
| αyy |
125.77 |
| αxz |
-0.02 |
| αyz |
0.00 |
| αzz |
70.94 |
| α0 |
3.1476x10-30esu |
| Hyperpolarizability ( β0)x10-30esu |
| βxxx |
1383.41 |
| βxxy |
211.90 |
| βxyy |
-151.05 |
| βyyy |
30.28 |
| βxxz |
0.10 |
| βxyz |
0.09 |
| βyyz |
-0.25 |
| βxzz |
-24.15 |
| βyzz |
-31.28 |
| βzzz |
0.19 |
| β0 |
10.595x10-30esu |
Standard value for urea (μ=1.3732 Debye, β0=0.3728x10-30esu)
Table 3: The NLO measurements of EMBU.
NBO analysis
The NBO analysis provides an efficient method for studying intraand inter-molecular bonding and interaction among bonds, and also provides a convenient basis for investigating charge transfer or conjugative interaction in molecular systems [31].
In this present study, the NBO analysis has been carried out with DFT/B3LYP/6-31G(d,p) level of basis set and which deals the intra-molecular charge transfer within the molecule. In any molecule, the π character of the bond plays an important role when compare with σ bond character. In such a way that this molecule delivers maximum delocalization energy during the transition between π and π* bond whereas the ED of the donor (Lewis) bond decreases with increasing of ED of acceptor (Non- Lewis) bonds. In our case, the conjugative π bonds in the phenyl ring shows maximum delocalization during the interaction with π* acceptor bonds. It is evident from our title compound that the energy transfer from πC1-C6 to π*C2-C3, πC4-C5 to π*C1-C6 and πC2-C3 to π*C11-N12 are reveals the hyperconjucative energy about 97.74, 92.51 and 86.69 KJ/mol, respectively. Similarly, the lone pair atoms such as oxygen and nitrogen also transfer more energy to donor and acceptor bonds. The LP(1)N14 to C13-O18 and LP(2)O18 to N12-C13 bonds transfer the energy about 252.71 and 106.78 KJ/mol, respectively and are listed in Table 4. The maximum hyperconjugative E(2) energy of lone pair atoms during the intra-molecular interaction, which leads the molecule towards medicinal and biological applications.
| Type |
Donor NBO (i) |
ED/e |
Acceptor NBO (j) |
ED/e |
E(2)
KJ/mol |
E(j)-E(i)
a.u. |
F(i,j)
a.u. |
| π -π* |
BD (2) C1 - C6 |
1.63787 |
BD*(2) C2 - C3 |
0.37327 |
97.74 |
0.28 |
0.072 |
| |
|
|
BD*(2) C4 - C5 |
0.26902 |
69.12 |
0.29 |
0.063 |
| |
|
|
BD*(1) C19 - H20 |
0.0084 |
10.96 |
0.65 |
0.041 |
| |
|
|
BD*(1) C19 - H22 |
0.0083 |
10.75 |
0.65 |
0.04 |
| π -π* |
BD (2) C2 - C3 |
1.62997 |
BD*(2) C1 - C6 |
0.32641 |
73.43 |
0.29 |
0.064 |
| |
|
|
BD*(2) C4 - C5 |
0.26902 |
81.46 |
0.29 |
0.068 |
| |
|
|
BD*(2) C11 - N12 |
0.13157 |
86.69 |
0.28 |
0.072 |
| π -π* |
BD (2) C4 - C5 |
1.67949 |
BD*(2) C1 - C6 |
0.32641 |
92.51 |
0.29 |
0.071 |
| |
|
|
BD*(2) C2 - C3 |
0.37327 |
75.06 |
0.28 |
0.065 |
| π -π* |
BD (2) C11 - N12 |
1.88985 |
BD*(2) C2 - C3 |
0.37327 |
32.64 |
0.35 |
0.05 |
| |
|
|
BD*(2) C13 - O18 |
0.32592 |
86.02 |
0.35 |
0.08 |
| π -π* |
BD (2) C13 - O18 |
1.99066 |
BD*(2) C11 - N12 |
0.13157 |
8.54 |
0.38 |
0.026 |
| |
|
|
BD*(2) C13 - O18 |
0.32592 |
6.86 |
0.38 |
0.024 |
| n -σ* |
LP (1) N12 |
1.9205 |
BD*(1) C3 - C11 |
0.03167 |
7.7 |
0.85 |
0.036 |
| |
|
|
BD*(1) C11 - H15 |
0.04014 |
44.06 |
0.77 |
0.081 |
| |
|
|
BD*(1) C13 - N14 |
0.06619 |
8.41 |
0.84 |
0.037 |
| |
|
|
BD*(1) C13 - O18 |
0.02348 |
38.07 |
0.99 |
0.086 |
| n -π* |
LP (1) N14 |
1.74159 |
BD*(2) C13 - O18 |
0.32592 |
252.71 |
0.28 |
0.119 |
| n -σ* |
LP (1) O18 |
1.97754 |
BD*(1) N12 - C13 |
0.08854 |
9.79 |
1.07 |
0.045 |
| |
|
|
BD*(1) C13 - N14 |
0.06619 |
8.54 |
1.15 |
0.044 |
| n -σ* |
LP (2) O18 |
1.8497 |
BD*(1) N12 - C13 |
0.08854 |
106.78 |
0.63 |
0.115 |
| |
|
|
BD*(1) C13 - N14 |
0.06619 |
97.61 |
0.71 |
0.117 |
*E(2) means energy of hyperconjucative interactions,
LP = Lone pair (non-bonding molecular orbital)
Table 4: The NBO analysis of EMBU.
HOMO-LUMO analysis
The HOMO and the LUMO are called frontier molecular orbitals as they lie at the outermost boundaries of the electrons of the molecules. The HOMO and LUMO are the main orbitals responsible for chemical stability. The HOMO–LUMO orbitals of the EMBU are as shown in Figure 5. The calculated values of the HOMO and LUMO energies and band gap energy are listed in Table 5. The positive and negative phases are represented in green and red color, respectively. HOMO represents the electrondonating ability of a molecule, whereas LUMO indicates its ability to accept electrons. The frontier orbital gap helps to characterize the chemical reactivity and kinetic stability of the molecule. In the present study, the energy gap have been calculated using B3LYP/6-31G(d,p) level are 4.76415 eV. This small energy gap is associated with high chemical reactivity and low kinetic stability.
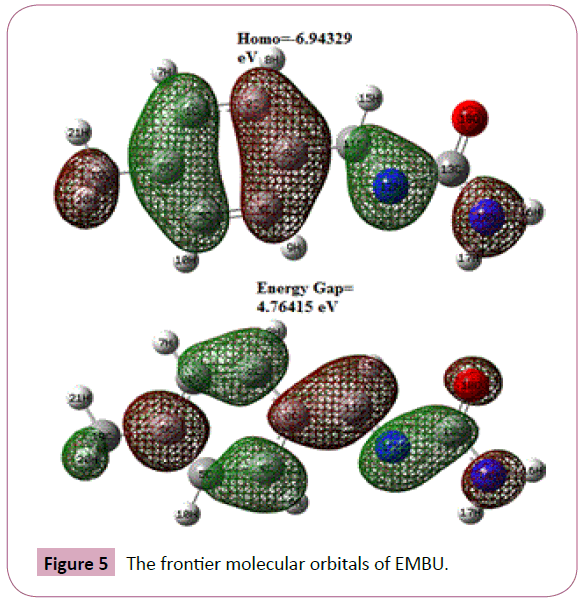
Figure 5: The frontier molecular orbitals of EMBU.
| Orbitals |
Energy (a.u) |
Energy (eV) |
| 39O |
-0.30489 |
-8.29636 |
| 40O |
-0.279438 |
-7.60379 |
| 41O |
-0.272717 |
-7.4209 |
| 42O |
-0.258641 |
-7.03788 |
| 43O |
-0.255165 |
-6.94329 |
| 44V |
-0.080083 |
-2.17914 |
| 45V |
-0.029496 |
-0.80262 |
| 46V |
-0.012194 |
-0.33181 |
| 47V |
-0.006217 |
-0.16917 |
| 48V |
0.00296 |
0.080545 |
Table 5: The frontier molecular orbitals of EMBU.
MEP analysis
The MEP surface map was calculated at B3LYP/6-31G(d,p) method. MEP is related to the ED. It is very useful descriptor in understanding the sites for electrophilic attacks and nuleophilic reactions as well as hydrogen bonding interactions [32]. The importance of MEP lies in the fact that it simultaneously displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions in terms of colour grading as shown in Figure 6. Potential increases in the order red < orange < yellow < green < blue. The colour code of these maps, where blue indicates the strongest interaction and red indicates the strongest repulsion. In this study, the negative region is located over the carbonyl group and the positive region is located over Hydrogen atom in the imine linkage.
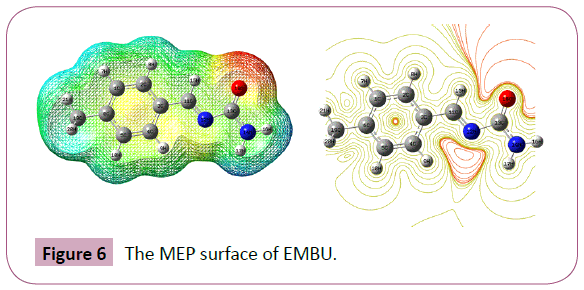
Figure 6: The MEP surface of EMBU.
Mulliken charges
Mulliken populations can be used to characterize the electronic charge distribution in a molecule and the bonding, anti-bonding, or non-bonding nature of the MOs for pair of atoms [33]. Mulliken atomic charge calculation has an important role in the application of quantum chemical calculation to molecular system. The total atomic charges of EMBU are calculated by Mulliken population analysis with DFT method B3LYP/6-31G(d,p) basis set are listed in Table 6. The Mulliken atomic charge plot for EMBU is plotted in Figure 7. The most positive charges calculated at C3/0.636162 and C6/0.651105 in the present molecule. The C6 atom has high positive charge due to the attachment of methyl group. Similarly, the negative charges were calculated at C1/-0.764424 and C19/-0.612242, respectively. From the results, C1 atom shows the higher negative charge due to the presence aromatic ring. The nitrogen and oxygen atoms also be a negative charge such as N12/-0.118445, N14/- 0.393447 and O18/-0.362573, respectively.
| Atoms |
Charges |
Atoms |
Charges |
| 1C |
-0.764424 |
12N |
-0.118445 |
| 2C |
-0.567188 |
13C |
0.247575 |
| 3C |
0.636162 |
14N |
-0.393447 |
| 4C |
-0.272493 |
15H |
0.187019 |
| 5C |
-0.251654 |
16H |
0.30635 |
| 6C |
0.651105 |
17H |
0.266017 |
| 7H |
0.138265 |
18O |
-0.362573 |
| 8H |
0.161024 |
19C |
-0.612242 |
| 9H |
0.179329 |
20H |
0.159652 |
| 10H |
0.176258 |
21H |
0.152303 |
| 11C |
-0.077157 |
22H |
0.158565 |
Table 6: The Mulliken atomic charges of EMBU.
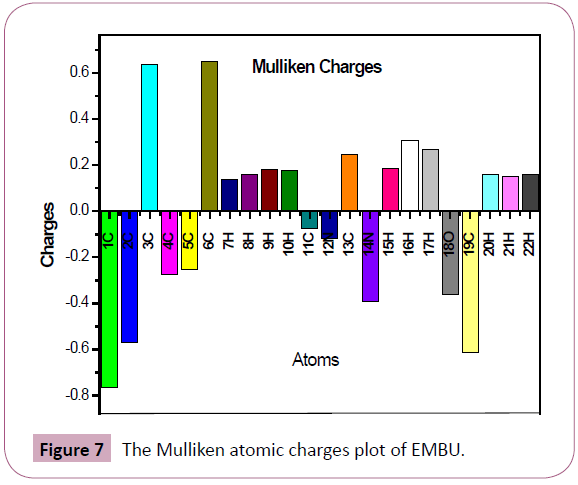
Figure 7: The Mulliken atomic charges plot of EMBU.
Thermodynamic properties
The standard thermodynamic functions: heat capacity (C0 p,m), entropy (S0 m) and enthalpy changes (H0 m) for the EMBU were calculated using DFT method at B3LYP/6-31G(d,p) basis set and are listed in Table 7. Therefore, it can be observed that these thermodynamic functions are increasing with temperature ranging from 100 to 1000 K due to the molecular vibrational intensities increase with temperature [34]. The correlation equations among thermodynamic functions due to the temperature were fitted by quadratic, linear and quadratic formula and the corresponding fitting factors (R2) for these thermodynamic properties are 0.99957, 0.99997 and 0.99971. The corresponding fitting equations are as follows, and the correlation graphs are shown in Figure 8.
| T |
S (J/mol.K) |
Cp (J/mol.K) |
ΔH (KJ/mol) |
| 100 |
322.65 |
102.31 |
7.77 |
| 200 |
431.05 |
157.53 |
21.14 |
| 300 |
504.31 |
248.38 |
39.40 |
| 400 |
572.12 |
314.48 |
68.66 |
| 500 |
656.22 |
361.16 |
102.77 |
| 600 |
746.02 |
414.85 |
129.31 |
| 700 |
811.21 |
450.54 |
171.54 |
| 800 |
852.00 |
459.45 |
237.19 |
| 900 |
928.71 |
498.26 |
285.37 |
| 1000 |
981.74 |
523.05 |
335.81 |
Table 7: Thermodynamic properties of EMBU at different temperatures.
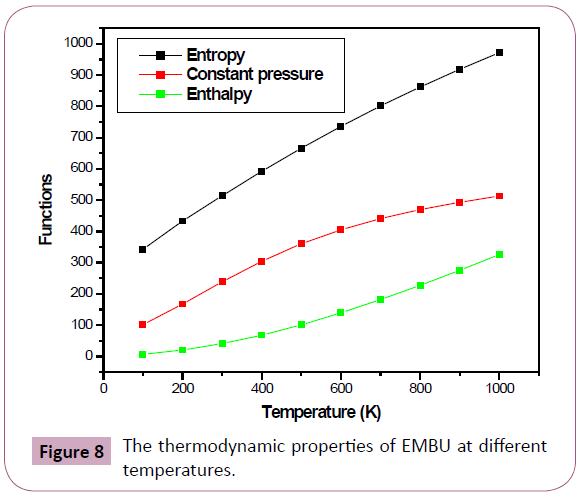
Figure 8: The thermodynamic properties of EMBU at different temperatures.
C0p,m = -1.73055 + 0.1542T – 6.3237x10-5 T2 (R2 = 0.99957)
S0m = 3.06457 + 0.13745T – 1.83581x10-5 T2 (R2 = 0.99997)
ΔH0m = 107.82213 + 0.00557T + 1.88572x10-5 T2 (R2 = 0.99971)
All the thermodynamic data are supportive information for further study on the molecule.
Conclusion
The quantum chemical calculations were performed at DFT method for the first time to the EMBU molecule. The bond parameters (bond lengths, bond angles and dihedral angles) are agree well with the related molecule XRD data. The observed and calculated wavenumbers of EMBU are well supported by the literature values. The hyperpolarizability (β0) value of EMBU molecule is calculated about 10.595x10-30 esu, which is twentyeight times greater than that of standard urea. The NBO analysis reveals that, the charge transfer occur within the molecule and the maximum energy takes place during π-π* transition. The energy gap of EMBU molecule is calculated at 4.76415 eV, which leads the title molecule to become less stable and more reactive. The reactive sites of EMBU molecule is predicted by MEP surface. In addition, Mulliken atomic charges and thermodynamic properties are also calculated and analyzed.
References
- Karthikeyan MS, Prasad DJ, Poojary B, Bhat KS, Holla BS, et al. (2006) Synthesis and biological activity of Schiff and Mannich bases bearing 2,4-dichloro-5-fluorophenyl moiety. Bioorg Med Chem 14: 7482-7489.
- Sriram D, Yogeeswari P, Sirisha N, Saraswat V (2006) Abacavir prodrugs: Microwave-assisted synthesis and their evaluation of anti-HIV activities. Bioorg Med. Chem Lett 16: 2127-2129.
- Liu G, Peiliao J, Huang S, Lishen G, Qinyu R (2001) Fluorescence Spectral Study of Interaction of Water-soluble Metal Complexes of Schiff-base and DNA. Anal Sci 17: 1031-1035.
- Khalaji AD, Hadadzadeh H, Gotoh K, Ishida H (2009) catena-Poly [[[N,N′-bis(3-methoxybenzylidene)ethylenediamine]copper(I)]-μ-thiocyanato-κ2N:S]. Acta Cryst 65: 70.
- Morshedi M, Amirnasr M, Triki S, Khalaji AD (2009) New (NS)2 Schiff base with a flexible spacer: Synthesis and structural characterization of its first coordination polymer [Cu2(μ-I)2(μ-(thio)2dapte)]n (1). Inorg. Chim Acta 362: 1637-1640.
- Sacconi L, Bertini I (1966) Complexes of Copper(II) with Schiff Bases Formed from Salicylaldehydes and N-Substituted Ethylenediamines. Inorg Chem 5: 1520-1522.
- Sales ZS, Mani NS (2009) An Efficient Intramolecular 1,3-Dipolar Cycloaddition Involving 2-(1,2 Dichlorovinyloxy)aryldiazomethanes: A One-Pot Synthesis of Benzofuropyrazoles from Salicylaldehydes J Org Chem 74: 891-894.
- Wang PH, Keck JG, Lien EJ, Lai MMC (1990) Design, synthesis, testing, and quantitative structure-activity relationship analysis of substituted salicylaldehyde Schiff bases of 1-amino-3-hydroxyguanidine tosylate as new antiviral agents against coronavirus. J Med Chem 33: 608-614.
- Chinigo GM, Paige M, Grindrod S, Hamel E, Dakshanamurthy S, et al. (2008) Asymmetric synthesis of 2,3-dihydro-2-arylquinazolin -4-ones: methodology and application to a potent fluorescent tubulin inhibitor with anticancer activity. J Med Chem 51: 4620-4631.
- Abd-Elzaher MM, Moustafa SA, Labib AA, Ali MM (2010) Synthesis, characterization and anticancer properties of ferrocenyl complexes containing a salicylaldehyde moiety. Monatsh Chem Chem Mon 141: 387-393.
- Prisakar VI, Tsapkov VI, Buracheéva SA, Byrké MS, Gulya AP (2005) Synthesis and Antimicrobial Activity of Coordination Compounds of Copper with Substituted Salicylaldehyde Thiosemicarbazones. J Pharm Chem 39: 313-315.
- Lazzarato L, Donnola M, Rolando B, Marini E, Cena C, et al. (2008) Searching for new NO-donor aspirin-like molecules: a new class of nitrooxy-acyl derivatives of salicylic acid. J Med Chem 51: 1894-1903.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (2004) Theoretical and Computational Aspects of Magnetic Organic Molecules. Gaussian Inc, Wallingford, USA.
- Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J Comput Chem 3: 214-218.
- Jamróz MH (2006) Vibrational modes of 2,6-, 2,7-, and 2,3-diisopropyl naphthalene. A DFT study. J Mol Struct 787: 172-183.
- Michalska D (2003) Raint Program, Wroclaw University of Technology, Poland.
- Michalska D, Wysokinski R (2005) The prediction of Raman spectra of platinum(II) anticancer drugs by density functional theory. Chem Phys Lett 403: 211-217.
- Ramesh Babu N, Subashchandrabose S, Syed Ali Padusha M, Saleem H, Erdogdu Y (2014) Synthesis and spectral characterization of hydrazone derivative of furfural using experimental and DFT methods. Spectrochim Acta A 120: 314-322.
- Song MZ, Fan CG (2009) (E)-N-(2-Furylmethylene)benzohydrazide. Acta Cryst E 65: o2800.
- Araujo-Andrade C, Giuliano BM, Gómez-Zavaglia A, Fausto R (2012) Structure and UV-induced photochemistry of 2-furaldehyde dimethylhydrazone isolated in rare gas matrices. Spectrochim Acta A 97: 830-837.
- Radom L, John A, Pople (1970) Molecular orbital theory of the electronic structure of organic compounds. IV. Internal rotation in hydrocarbons using a minimal Slater-type basis. J American Chem Soc 92: 4786-4795.
- Pople JA, Scott AP, Wong MW, Radom L (1993) Scaling Factors for Obtaining Fundamental Vibrational Frequencies and Zero-Point Energies from HF/6-31G* and MP2/6-31G* Harmonic Frequencies. Israel J Chem 33: 345-350.
- Dabbagh HA, Teimouri A, Chermahini AN, Shahraki M (2008) DFT and ab initio study of structure of dyes derived from 2-hydroxy and 2,4-dihydroxy benzoic acids. Spectrochim Acta A 69: 449-459.
- Varasanyi G (1974) Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives, vol. 1–2, Adam Hilger.
- Ravikumar C, Hubert Joe H, Jayakumar VS (2008) Charge transfer interactions and nonlinear optical properties of push–pull chromophore benzaldehyde phenylhydrazone: A vibrational approach. Chem Phy Lett 460: 552-558.
- Rastogi V, Palafox MA, Tanwar RP, Mittal L (2002) 3,5-Difluorobenzonitrile: ab initio calculations, FT-IR and Raman spectra. Spectrochim. Acta A 58: 1987-2004.
- Silverstein M, Basseller CG, Morill C (1981) Spectrometric identification of organic compound, JohnWiley; Network.
- Lorenc J (2012) Dimeric structure and hydrogen bonds in 2-N-ethylamino-5-metyl-4-nitro-pyridine studied by XRD, IR and Raman methods and DFT calculations. Vib Spectrosc 61: 112-123.
- Palafox MA, Nunez JL, Gil M (2002) Accurate scaling of the vibrational spectra of aniline and several derivatives. J Mol Struct 593: 101-106.
- Silverstein M, Clayton Basseler G, Morill C (1981) Spectrometric Identification of Organic Compounds, Wiley, New York.
- Snehalatha M, Ravikumar C, Hubert Joe I, Sekar N, Jayakumar VS (2009) Spectroscopic analysis and DFT calculations of a food additive Carmoisine. Spectrochim Acta A 72: 654-662.
- Politzer P, Truhlar DG (1981) Chemical Applications of Atomic and Molecular Electrostatic Potentials.
- Mulliken RS (1955) Electronic Population Analysis on LCAO–MO Molecular Wave Functions. J Chem Phys 23: 1833-1841.
- Sajan D, Lynnette J, Vijayan N, Karabacak M (2011) Natural bond orbital analysis, electronic structure, non-linear properties and vibrational spectral analysis of l-histidinium bromide monohydrate: A density functional theory. Spectrochim Acta A 81: 85-98.