Yu-Shi Tian1, Norihito Kawashita2,3, Masanori Kameoka4and Tatsuya Takagi2,3
1Graduate School of Information Sciences and Technology, Osaka University, 1-5 Yamadaoka, Suita, Osaka 565-0871, Japan
2Graduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
3Research Institute for Microbial Diseases, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
4Department of International Health, Kobe University Graduate School of Health Sciences, 7-10-2 Tomogaoka, Suma-Ku, Kobe 654-0142, Japan
Corresponding Author:
Tatsuya Takagi
Graduate School of Pharmaceutical Sciences
Osaka University, 1-6 Yamadaoka, Suita
Osaka 565-0871, Japan
Tel: +81 6 68798243
Fax: +81 6 68798242
Email: satan@gen-info.osaka-u.ac.jp
Received date: September 27, 2016; Accepted date: November 08, 2016; Published date: November 15, 2016
Citation: Tian YS, Kawashita N, Kameoka M, et al. Novel Anti-Human Immunodeficiency Virus Compounds with Activity against Cyclophilin A: A Look Back. J Prev Inf Cntrl. 2016, 2:2.
Copyright: © 2016 Tian YS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords
HIV; CypA; Inhibitor; Antiviral drugs.
Introduction
The history of human immunodeficiency virus (HIV) has seen remarkably change in a relatively short period of time due to the global increase of anti-retroviral therapy coverage. There is a common worldwide goal to eliminate HIV-related mortality and to end the AIDS pandemic by the year 2030. With great excitement, we appear to be continuously moving toward this target. However, according to the statistics from Global AIDS Update 2016, much more work remains to be done, because approximately 2.1 million individuals were newly infected with HIV in 2015 and the number of people living with HIV worldwide has reached up to 36.7 million [1]. Nevertheless, the development, approval, and clinical use of anti-retroviral drugs have shown encouraging results in the fight against this fatal virus, bringing much hope to HIV patients.
To date, numerous researchers and multiple pharmaceutical manufacturers have focused their efforts on searching for and developing active compounds to combat HIV. Currently, a total of 39 anti-HIV drugs (Figure 1) have been approved by the US Food and Drug Administration, which are classified into nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, protease inhibitors, fusion inhibitor, entry inhibitors-CCR5 co-receptor antagonist, HIV integrase strand transfer inhibitors, and multi-class combination products [2].
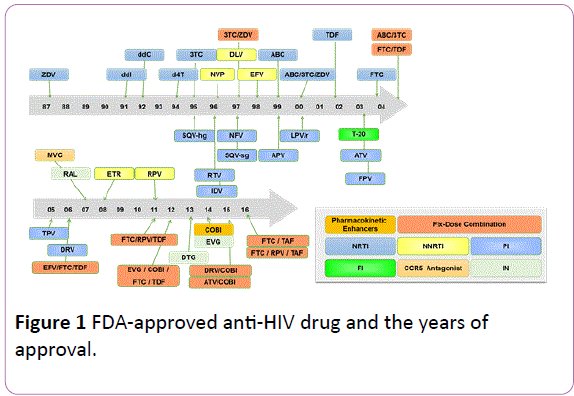
Figure 1: FDA-approved anti-HIV drug and the years of approval.
The combined use of these drugs, also known as the antiviral cocktail regimen or highly active antiretroviral therapy (HAART/ Highly Active ART), has succeeded in reducing the viral loads and is very effective. Compared to the single use of an antiviral drug, HAART effectively controls the emergence rate of mutants at a relatively low level; however, drug resistance remains a significant obstacle to successful treatment. Therefore, novel drugs, especially those belonging to novel classes, are continuously sought. Cyclophilin A (CypA), a host factor is recently focused on. Both circle peptides and small molecules were designed to inhibit this target. In this review, we take a retrospective look at research related to CypA as a new anti-HIV target, and the identification of candidate inhibitors.
CypA Structure and Its Possible Function
CypA is a target of the immunosuppressant drug cyclosporine A (CsA) (Figuire 2) and was also found to bind to the HIV capsid protein (CA) [3, 4]. These observations stimulated several studies on the functional significance of CypA. One particularly important aspect is to elucidate the role of this host factor during viral infection or proliferation. Resolving this question may reveal CypA as a suitable target for novel drug design and also help to acquire more information on information on the life cycle of HIV. Although many independent experiments have been carried out to address this question, the significance of CypA for HIV is not yet fully understood.
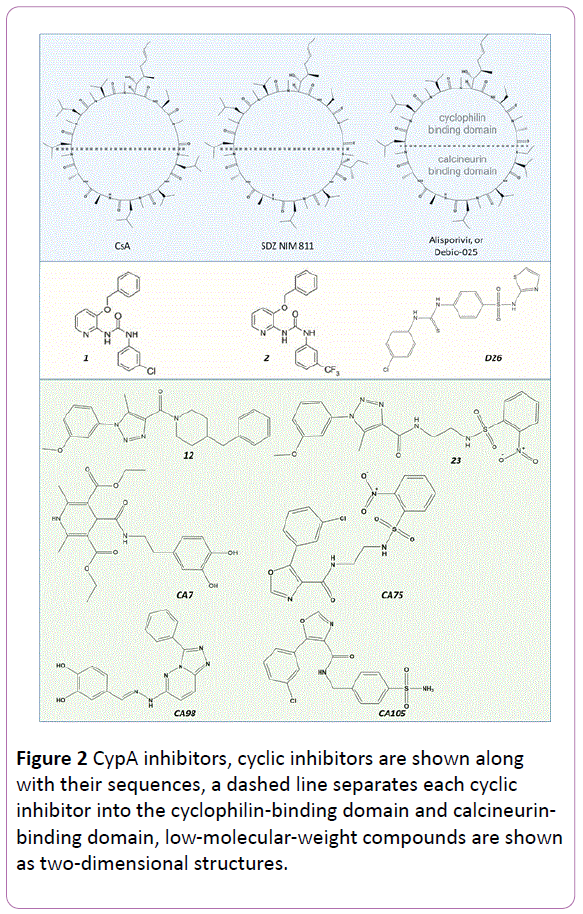
Figure 2: CypA inhibitors, cyclic inhibitors are shown along with their sequences, a dashed line separates each cyclic inhibitor into the cyclophilin-binding domain and calcineurinbinding domain, low-molecular-weight compounds are shown as two-dimensional structures.
The HIV contains a viral genetic core (double-stranded RNA) and necessary enzymes for viral infection and proliferation in a cone-shaped form. The components are hexamers and pentamers of two domains (an N-terminal domain [NTD] and a C-terminal domain [CTD]) linked by a linker structure. HIV infections begin with recognition of host target cells, followed by insertion of this parachute into the cytosol of target cells. Subsequently, the digestion of this core triggers exposure of the viral genome. Finally, the integration into host DNA conquers the target cell and the virus begins to produce its own descendants. The exact site of CA-CypA binding has been examined by several groups. Structural resolution and genetic mutation approaches have clarified that residues G89-P90 of the NTD of CA are the target of the CypA enzyme center. To obtain a glance of the structure, we superimposed two published X-ray crystallography structures, namely 2CYH [5] and 1AK4 [6] (Figure 3). The former structure is a co-crystal of CypA and a dipeptide (AP), and the latter is a complex of CypA and part of CA. The two CypA backbones are almost identical and show an 8-strand beta barrel structure. Moreover, both the dipeptide AP from 2CYH and the "HAGPI" peptide of CA from 1AK4 were found to be inserted into the same region of CypA, which is also the target of CsA. This pocket is the center for peptidylprolyl cis-trans isomerase activity, and it is assumed that the isomerization of G89-P90 is catalyzed by CypA [7-9].
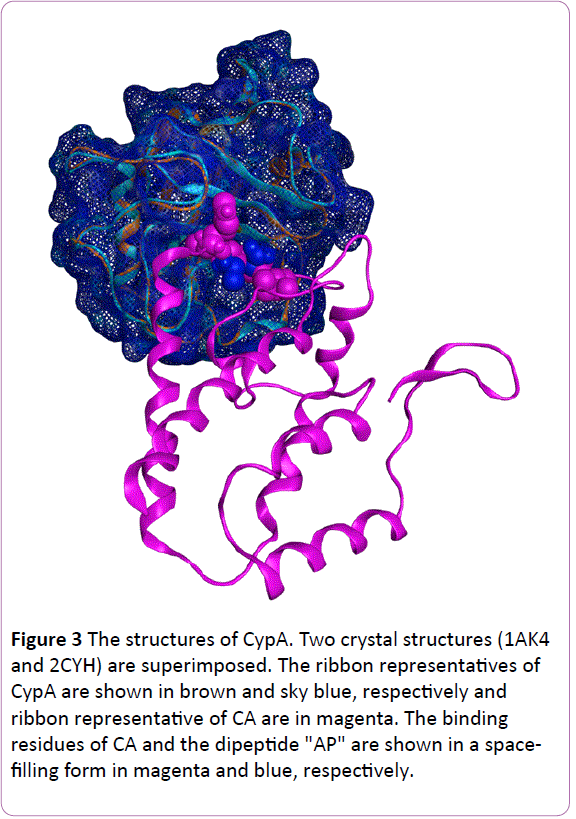
Figure 3: The structures of CypA. Two crystal structures (1AK4 and 2CYH) are superimposed. The ribbon representatives of CypA are shown in brown and sky blue, respectively and ribbon representative of CA are in magenta. The binding residues of CA and the dipeptide "AP" are shown in a spacefilling form in magenta and blue, respectively.
Another observation is that, unlike other cyclophilins, CypA is packaged into virions, which is also a specific feature of HIV-1, and is not found in simian immunodeficiency virus (SIV) or HIV-2. Sequence mutation analysis showed that introducing the residues from position 86 to 93 of HIV-1 p24 can make the SIVmac virion incorporate CypA, resulting in sensitivity to CsA [10], further revealing the CypA-binding site of CA. Given that the host CypA is packaged into the core during viral assembly and interacts with CA, it was first assumed that CypA plays a role in viral producer cells. However, a study using owl monkey cells and CypA virions showed no obvious change in the infectious strength in the presence or absence of CypA [11]. This suggests that the CypA-CA interaction may act to disassemble CA and/or be part of the transport step of the viral core in target cells. The same study showed that the CypA-CA interaction may be a mechanism to escape from the host restriction factor Ref-1 in the target cell, and that CsA inhibits that action [11].
Besides Ref-1, CypA was demonstrated to interact with nucleopore protein complex (NPC) and other factors that perform the uncoating reaction and/or transportation to the nucleus (Figure 4). Pre-integration complex (PIC) interacts with several factors, including cleavage and polyadenylation specific factor 6 (CPSF6) and transportin SR2 (TRN-SR2)/transportin 3 (TNPO3). CypA is suggested to be a regulator of these factors both directly and indirectly. The function of CypA in viral infection was also reviewed in detail by Hopkins and Gallay [12]; readers can obtain more information from their review or refer to the original published works cited within.
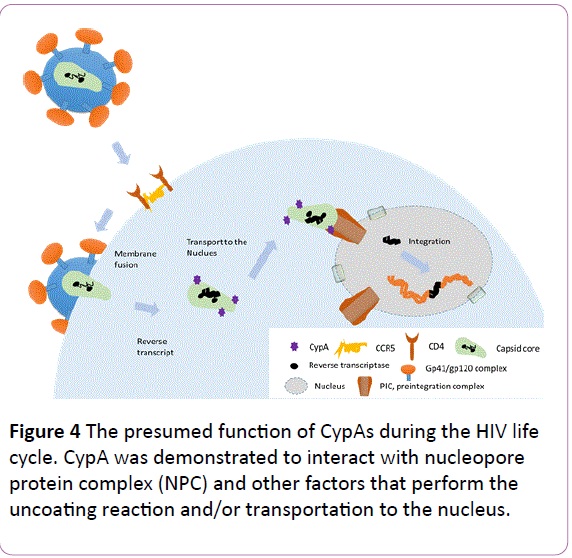
Figure 4: The presumed function of CypAs during the HIV life cycle. CypA was demonstrated to interact with nucleopore protein complex (NPC) and other factors that perform the uncoating reaction and/or transportation to the nucleus.
Approaches to Inhibit the Host Factor CypA
To our knowledge, CsA, a peptide ring of 11 amino acids (Figure 2), was first found to inhibit HIV-1 in 1988 [13], well before the emergence of studies on the CypA-CA interaction. Because HIV was not well understood at that time, the effects of CsA as an anti-HIV agent were directly tested in patients. Unfortunately, the clinical results showed worse laboratory values, and therefore CsA was proven to be not applicable as an anti-HIV drug. Now, we know that CsA is an immunosuppressive drug, which is used to regulate allograft rejection via forming a triad complex with calcineurin and the consequent activation of the nuclear factor of activated T-cells pathway. The mechanism is independent of the CsA-CypA-CA interactions observed in the context of HIV-related function. Therefore, if CypA inhibitors can be designed for use in HIV treatments, their immunosuppressive potential must be eliminated. The 11 residues can be separated into a CypA-binding domain and a calcineurin-binding domain. Modifications of the residues in calcineurin-binding domain have been carried out to eliminate ligand’s immunosuppressive potential. For example, altering the Ile residue to MeIle at position 4 hindered the binding of calcineurin and changed CsA into a non-immunosuppressive agent (SDZ NIM 811) (Figure 2). However, this molecule was found to work better against hepatitis C virus (HCV) [14, 15]. The combination of SDZ NIM 811 and PEG-IFN/ribavirin as an anti-HCV drug has been tested in clinical trials [16] and thrombocytopenia was observed as an adverse event. Although at a dose of 75 mg/day the hepatocytoprotective effect can be detected, due to the less active compared to Alisporivir or Debio-025, the clinical trials were terminated [17]. Debio-025 was another modification reported as a more potent and less toxic molecule compared to CsA with a selectivity index greater than 300 [18]. However, some clinical isolates showing a natural resistance to Debio-025 were reported [19]. Debio-025 is now in Phase II of a clinical trial as an anti-HCV drug. More recently, CPI-431-32 was reported as a dual inhibitor for both HIV and HCV [20]. This molecule was expected to be effective in HIV/HCV co-infected patients. Some other cyclic peptides inhibiting CypA have been reported according to structure–activity relationship (SAR) studies [21, 22]. Although several modifications of CsA have been successful, peptide drugs are usually hard to be administrated orally.
The need to search for small inhibitors or to shorten the peptides has been raised. During the investigations to determine the specific site of CA that CypA binds to, the peptide N-acetyl- Ala-Ala-Pro-Ala aminomethyl coumarin was identified [23, 24]. Subsequently, dipeptides (Ala-Pro, Gly-Pro, His-Pro, Ser-Pro) were co-crystalized with CypA and were found to bind to the site as competitive inhibitors [5, 25]. The inhibitory activities of a series of mimic peptides derived from CA were tested, and a short peptide with the sequence Dav-His-Ala-Gly-Pro-Ile-NHBn was found to show inhibitory activity at the micromolar scale [26]. Based on the co-crystal data of the CypA/CsA complex, a rational design of non-peptide compounds was carried out [27] using pharmacophores and docking studies, and two diaryl urea structures, namely 1, 2 (Figure 2), were found to show inhibitory isomerase activity, with a 50% inhibitory concentration (IC50) of around 0.3 μM. After careful analysis of the first-round hits and interactions of CsA with the receptor CypA, the authors also carried out SAR studies and found two compounds with IC50 values of 14 nM and 20 nM, respectively. After checking the infection and production of HIV-1, the authors found one active compound that could impair both infection and production. In 2009, Li et al. [28] tested thiourea compounds as dual inhibitors of CA and CypA with the aim of targeting both the CA proteins and CypA-CA interaction, and 15 compounds were identified as potent candidates (one of them named D26 is shown in Figure 2). Despite the numerous efforts put forth, so far no drug classified as a "CypA inhibitor" for HIV has yet been approved.
Our Approach to CypA Inhibitor Design
We carried out an in silico screen with a commercial lowmolecular- weight compound database from ChemGenesis (https://www.all-chemy.com) [29] to find a potent inhibitor against the CypA-CA interaction. The receptor adopted was 2CYH, because it does not show any obvious difference with other structures reported for CypA, as shown in Figure 3 and we could successfully re-dock the ligand back to the receptor. The molecules were not filtered because of the low amount of compounds available compared to other in silico studies. The detailed computational parameters can be found in another publication [29]. We first used both virus-infected CXCR4 and MT-4 cells to check the anti-HIV replication activities of the candidates selected, and then used virus-producing proviral DNA-transfected 293T cells, together with the CXCR4 and MT-4 cells, to confirm the effects on cell viability. We found two compounds (23 and 12, shown in Figure 2) that exhibited comparable activities to positive controls, and showed lower toxicity based on the observations of cell viability.
We subsequently repeated the experiments, added some other in silico screening results, and further checked whether these candidates show potential for inhibiting the CypA-CA interaction [30]. Because more compounds were tested compared to our first study, according to the criteria of greater activity, less toxicity, clearance of the solubility problem, and consistency in the results or reproducibility based on our experiments, we rechecked all of the candidates for further analysis. As a result, four compounds (CA7, CA75, CA98, and CA105, shown in Figure 2) were analyzed as candidates, together with CsA as a positive control. Although, as mentioned above, CypA in infected cells was more important according to others’ reports, our four selected compounds showed nearly no activity in infected cells (CA105 was sensitive in MT4 but not in U87 cells). However, viral replication in viral-producing cells was potentially inhibited by these four selected compounds that showed relatively low cytotoxicity. Therefore, we carried out further experiments using western blot analysis and assays with CypA-binding-deficient CA to confirm the target. To our surprise, at least three of the four compounds did not appear to target CypA-CA binding in virus-producing cells. Nonetheless, in the presence of 30 μM CA75, the deficient mutations of the CypAbinding site in CA could rescue the viral replications to 45% and 41% in both MT4 and U87 cell lines, respectively. This result was difficult to interpret because incorporation of CypA into the HIV-1/VSV-G virions was not reduced by CA75. Thus, we assumed that CA75 interacts with CypA-CA binding in a dynamic state or not particularly strongly.
The results of in vitro experiments suggested that our in silico design might be incorrect. Although docking studies are widely used nowadays for drug design, false positive hits are sometimes obtained. There are multiple reasons for this. In some cases, the source (structure of targets) or the mechanism (docking software packages and scoring functions) might be the problem. However, in the majority of cases, the lower activities observed in vitro may be caused by the fact that we can only model the target and compounds without consideration of different cell systems or the interactions between other factors. To resolve this issue, we believe that conducting back-and-forth procedures between in silico and experimental studies should be carefully utilized.
Although the detailed mechanisms or targets of our four candidate compounds remain unclear, their properties may be acceptable to be further considered as anti-HIV targets in future studies.
Summary
In this review, we have looked back on the studies of the function of CypA in the HIV life cycle and the continued search for candidate inhibitors. Although the detailed role and mechanism of action of CypA are not yet understood, we have been able to select potent inhibitor candidates. We believe that more work in this direction will be able to push these candidates toward clinical studies, and eventually, novel drugs can be approved for people living with HIV.
Moreover, given that HIV or AIDS was long regarded as a fatal disease, and because the infectious route is sexually or bloodtransmitted, patients experienced or continue to experience discrimination, which has hindered their willingness to visit a hospital at an early stage of infection. However, with the accumulated knowledge of HIV, we have become more familiar with this virus. Currently, HIV can be well controlled using ART and people living with HIV can obtain a high quality of life equivalent to that of healthy people. In addition, several programs are available to increase the ART coverage for patients living in low-income countries. Therefore, the fight against HIV and AIDS is no longer a national project, but is a goal of international collaboration. Virologists are continuously promoting the eradication of this virus through their efforts in finding novel target(s) and new drugs.
Acknowledgement
None
Authors’ Contribution
Y.S.T. wrote the first draft of the article. All of the authors revised the manuscript and approved the final version.
References
- Global AIDS Update (2016) UNAIDS.
- FDA-Approved HIV Medicines. HIV/AIDS fact sheets. Education Materials.
- Franke EK, Yuan HEH, Luban J (1994) Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372: 359-362.
- Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP (1993) Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73: 1067-1078.
- Zhao Y, Ke H (1996) Mechanistic implication of crystal structures of the cyclophilin-dipeptide complexes. Biochemistry 35: 7362-7368.
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, et al. (1996) Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87: 1285-1294.
- Braaten D, Aberham C, Franke EK, Yin L, Phares W, et al. (1996) Cyclosporine A-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes the functional target of cyclophilin A. J Virol 70: 5170-5176.
- Colgan J, Yuan HE, Franke EK, Luban J (1996) Binding of the human immunodeficiency virus type 1 Gag polyprotein to cyclophilin A is mediated by the central region of capsid and requires Gag dimerization. J Virol 70: 4299-4310.
- Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, et al. (1997) Molecular recognition in the HIV-1 capsid/cyclophilin A complex 1. J Mol Biol 269: 780-795.
- Bukovsky AA, Weimann A, Accola MA, Göttlinger HG (1997) Transfer of the HIV-1 cyclophilin-binding site to simian immunodeficiency virus from Macaca mulatta can confer both cyclosporin sensitivity and cyclosporin dependence. Proc Natl Acad Sci 94: 10943-10948.
- Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, et al. (2003) Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med 9: 1138-1143.
- Hopkins S, Gallay PA (2015) The role of immunophilins in viral infection. Biochim Biophys Acta BBA 1850: 2103-2110.
- Andrieu JM, Even P, Venet A, Tourani JM, Stern M, et al. (1988) Effects of cyclosporin on T-cell subsets in human immunodeficiency virus disease. Clin Immunol Immunopathol 1988; 47: 181-198.
- Tang H (2010) Cyclophilin inhibitors as a novel HCV therapy. Viruses 2: 1621-1634.
- Ma S, Boerner JE, TiongYip C, Weidmann B, Ryder NS, et al. (2006) NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob Agents Chemother 50: 2976-2982.
- Fischer G, Gallay P, Hopkins S (2010) Cyclophilin inhibitors for the treatment of HCV infection. Curr Opin Investig Drugs 11: 911-918.
- Flisiak R, Parfieniuk-Kowerda A (2012) Cyclophilin inhibitors. Curr Hepatitis Rep 11: 153-159.
- Daelemans D, Dumont JM, Rosenwirth B, De Clercq E, Pannecouque C (2010) Debio-025 inhibits HIV-1 by interfering with an early event in the replication cycle. Antiviral Res 85: 418-421.
- Gallay PA, Ptak RG, Bobardt MD, Dumont JM, Vuagniaux G, et al. (2013) Correlation of naturally occurring HIV-1 resistance to DEB025 with capsid amino acid polymorphisms. Viruses 5: 981-997.
- Gallay PA, Bobardt MD, Chatterji U, Trepanier DJ, Ure D, et al. (2015) The novel cyclophilin inhibitor CPI-431-32 concurrently blocks HCV and HIV-1 infections via a similar mechanism of action. Plos One 10: e0134707.
- Falck P, Guldseth H, Åsberg A, Midtvedt K, Reubsaet JLE (2007) Determination of ciclosporin A and its six main metabolites in isolated T-lymphocytes and whole blood using liquid chromatography-tandem mass spectrometry. J Chromatogr B 852: 345-352.
- Maurer G, Loosli HR, Schreier E, Keller B (1984) Disposition of cyclosporine in several animal species and man. I. Structural elucidation of its metabolites. Drug Metab Dispos 12: 120-126.
- Kallen J, Walkinshaw MD (1992) The X-ray structure of a tetrapeptide bound to the active site of human cyclophilin A. Febs Lett 300: 286-90.
- Kallen J, Spitzfaden C, Zurini MGM, Wider G, Widmer H, et al. (1991) Structure of human cyclophilin and its binding site for cyclosporin A determined by X-ray crystallography and NMR spectroscopy. Nature 353: 276-279.
- Ke H, Mayrose D, Cao W (1993) Crystal structure of cyclophilin A complexed with substrate Ala-Pro suggests a solvent-assisted mechanism of cis-trans isomerization. Proc Natl Acad Sci 90: 3324-3328.
- Li Q, Moutiez M, Charbonnier JB, Vaudry K, Ménez A, et al. (2000) Design of a Gag pentapeptide analogue that binds human cyclophilin A more efficiently than the entire capsid protein: New insights for the development of novel anti-HIV-1 drugs. J Med Chem 43: 1770-1779.
- Guichou JF, Viaud J, Mettling C, Subra G, Lin YL, et al. (2006) Structure-based design, synthesis and biological evaluation of novel inhibitors of human cyclophilin A. J Med Chem 49: 900-910.
- Li J, Tan Z, Tang S, Hewlett I, Pang R, et al. (2009) Discovery of dual inhibitors targeting both HIV-1 capsid and human cyclophilin A to inhibit the assembly and uncoating of the viral capsid. Bioorg Med Chem 17: 3177-3188.
- Tian YS, Verathamjamras C, Kawashita N, Okamoto K, Yasunaga T, et al. (2013) Discovery of novel low-molecular-weight HIV-1 inhibitors interacting with cyclophilin A using in silico screening and biological evaluations. J Mol Model 19: 465-475.
- Verathamjamras C, Tian YS, Kawashita N, Okamoto K, Yasunaga T, et al. (2015) Search for low molecular weight compounds that inhibit human immunodeficiency virus type 1 replication. J Infect Dis Ther 3: 1000218.