Keywords
Golgi; Tumor; Myosin; Apoptosis; Carcinogenesis
Golgi Disorganization and the ‘Glycosylation Signature’ of Cancer
Mammalian Golgi is the central station of glycosylation, composed of more than 250 glycosyltransferases that are highly organized according to the biosynthetic steps in which they participate [1]. Not surprisingly, perturbation in Golgi morphology leads to reordering of these enzymes, which in turn results in the formation of specific glycosyl epitopes. The well-recognized abnormal glycosylation in cancer occurs in the increase of sialylation, associated with a metastatic cell phenotype that has been detected both in clinical settings and experimental models [2]. It is a widely accepted view that overexpression of different sialylated antigens has not only a significant correlation with tumor progression, but that it also can protect cancer cells from apoptosis and has been suggested to confer resistance to therapy [3-6]. The major breakthrough in cancer glycobiology has come from pioneering experiments which have shown that inhibition of N- or O-glycan sialylation reduces the metastatic potential of colon cancer cells [7-9], the fragmented Golgi phenotype of which was later frequently reported on [10-13].
One of the most studied tumor-associated carbohydrate antigens is the Tn antigen, an initial O-glycan formed by linking GalNAc to the protein at Ser or Thr residue (Figure 1a). The Tn antigen can be further converted to the Core 1 structure (T antigen) by β1,3 galactose extension, the reaction catalyzed by Core 1 synthase (C1GalT1). Importantly, the α6-sialyltransferases (ST6GalNAc) compete with C1GalT1 for GalNAc substrate and represent an alternative short pathway which results in the formation of sialyl-Tn antigen (STn) (Figure 1a). Overexpression of Tn and STn antigens was described in breast, pancreas, stomach, lung, bladder, and uterus adenocarcinoma [14-17]. Due to its simple structure, Tn antigen was successfully targeted in clinics by the use of several therapeutic vaccines [18-20]. However, enthusiasm for this treatment diminished when prostate cancer treated with Tn-based vaccines did not bear the expected fruits and led to an equivocal conclusion [21]. This result also agreed with a publication which described only 20% of prostate tumors as Tn positive [22]. Further, an anti-cancer vaccine, Theratope, designed to address the STn epitope, failed on phase III of its clinical trial [23].
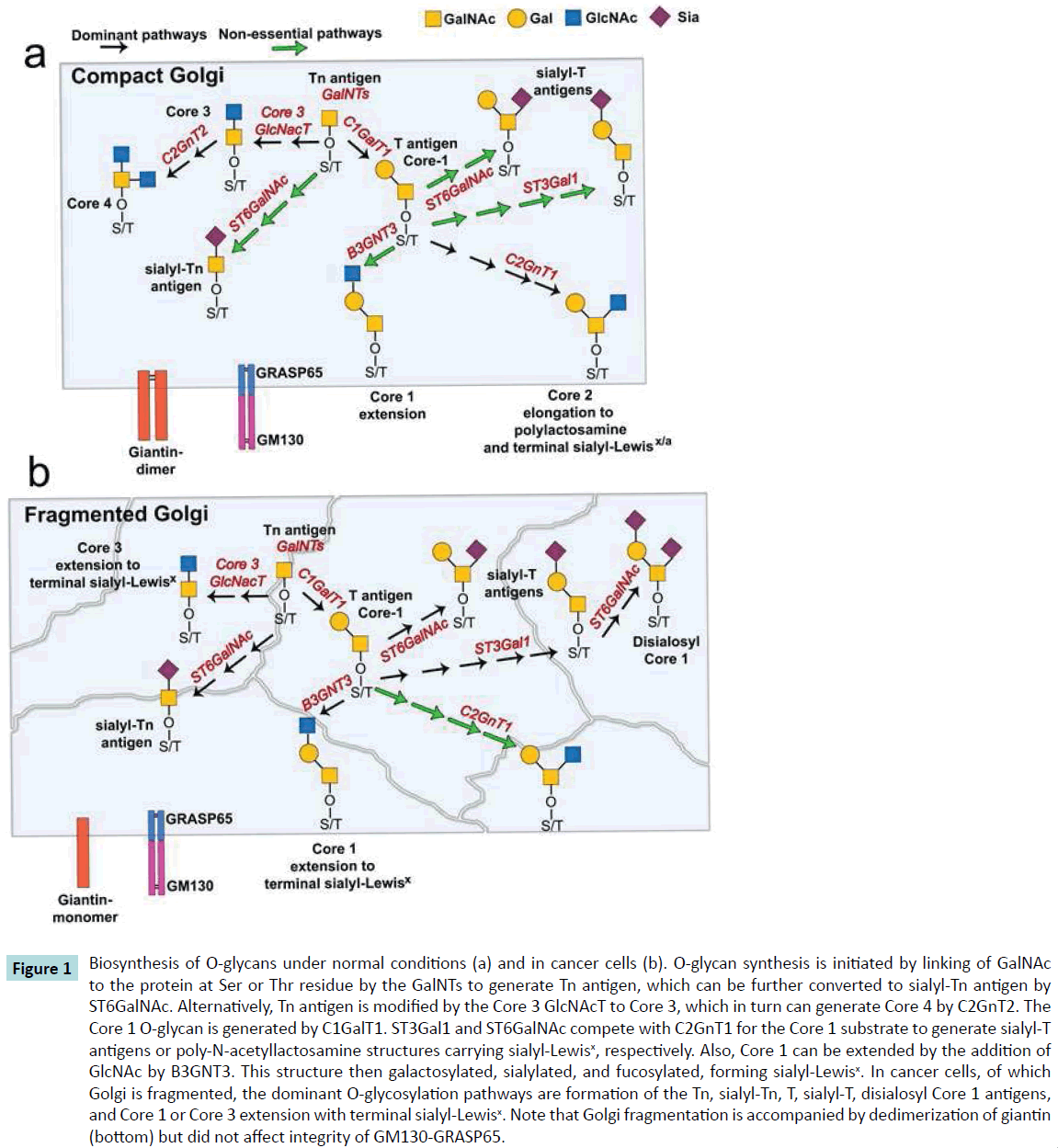
Figure 1 Biosynthesis of O-glycans under normal conditions (a) and in cancer cells (b). O-glycan synthesis is initiated by linking of GalNAc to the protein at Ser or Thr residue by the GalNTs to generate Tn antigen, which can be further converted to sialyl-Tn antigen by ST6GalNAc. Alternatively, Tn antigen is modified by the Core 3 GlcNAcT to Core 3, which in turn can generate Core 4 by C2GnT2. The Core 1 O-glycan is generated by C1GalT1. ST3Gal1 and ST6GalNAc compete with C2GnT1 for the Core 1 substrate to generate sialyl-T antigens or poly-N-acetyllactosamine structures carrying sialyl-Lewisx, respectively. Also, Core 1 can be extended by the addition of GlcNAc by B3GNT3. This structure then galactosylated, sialylated, and fucosylated, forming sialyl-Lewisx. In cancer cells, of which Golgi is fragmented, the dominant O-glycosylation pathways are formation of the Tn, sialyl-Tn, T, sialyl-T, disialosyl Core 1 antigens, and Core 1 or Core 3 extension with terminal sialyl-Lewisx. Note that Golgi fragmentation is accompanied by dedimerization of giantin (bottom) but did not affect integrity of GM130-GRASP65.
In the meantime, growing evidence in the literature has indicated that the increased expression of other tumor-associated carbohydrate antigens is achieved by the extension of Core 1 structure [24,25]. We know that T antigen consists of unsialylated Core 1 structures, but Core 1 can be converted to sialyl-T antigens by α3-sialyltransferases (ST3Gal1) or by ST6GalNAc1. Notably, both T and sialyl-T antigens are overexpressed in colon, breast and prostate cancer [26-29], but they are predominantly synthesized in the absence of the active Core 2 extension enzyme, C2GnT1 [30-34]. In normal prostate and androgen-refractory prostate cancer cells, C2GnT1 was detected in the Golgi and its function seen to result in a synthesis of polylactosamine, which makes these cells susceptible to galectin-1-induced apoptosis [34,35]. In advanced androgen-refractory prostate cancer cells as well as in primary prostate tumors, Golgi was found to be fragmented. The level of Golgi disassembly was correlated with Gleason score and metastasis, but most importantly, C2GnT1, contrary to the ST3Gal1, was mislocalized to the endoplasmic reticulum (ER). Consequently, the expression level of polylactosamine was reduced, while sialyl-T antigen was enhanced, allowing cells to evade galectin-1-induced apoptosis [34] (Figure 1b).
Understanding the phenomenon of mislocalization of Golgi residential enzymes in the cells with fragmented Golgi phenotype was arrived at with published research that shed light on the features of the coiled-coil-rich proteins associated with the Golgi matrix, including golgins and Golgi ReAssembly Stacking Proteins (GRASPs). Central to their function are: a) the building blocks of the Golgi architecture; b) the template for Golgi reassembly; and c) the docking sites for the transport vesicles that carry various cargoes and residential proteins, including glycosidases and glycosyltransferases [36-40]. Intriguingly, knockdown of golgins by siRNAs results in a reorganization of Golgi in the majority of cases. However, most crucial Golgi disassembly was detected in cells lacking giantin [34,41,42].
Giantin is the highest molecular weight (376 kDa) Golgi matrix protein. It consists of a short C-terminal domain located in the Golgi lumen [43], where a disulfide bond connects two monomers to form an active homodimer, followed by a one-pass transmembrane domain and then a large (≥350 kDa) N-terminal region [44,45]. It has also been shown that fragmentation of Golgi in advanced prostate cancer is accompanied by impaired dimerization of giantin [34]. Given that giantin is essential for cross-bridging cisternae during Golgi biogenesis [34,44], it is becoming clear that giantin dedimerization may cause fragility of the Golgi structure. In addition to giantin, GM130 and GRASP65 are also regulating initial steps of O-glycosylation. In the cells with normal, compact Golgi, C2GnT1 is docked to this organelle by a giantin-dependent mechanism, whereas C1GalT1 and ST3Gal1 use both giantin and GM130-GRASP65 [40]. It is worth noting that GM130 is able to make a complex with giantin in the absence of GRASP65 [40,46]. This alternative docking mechanism allows C1GalT1 and ST3Gal1 (but not C2GnT-L) to be delivered to the Golgi, even though giantin is missed or presented as a monomer and Golgi is fragmented (Figure 1b). Finally, the shift from the Core 2 pathway to ST3Gal1-mediated glycosylation provides an excessive expression of sialyl-T antigen [34].
What remain unclear are the details of the mechanism whereby the Golgi in some cancer cells prefers to form Tn and STn instead of T and sialyl-T antigens or Core 2 elongation. Different possibilities can be envisaged here. The first and simplest explanation of overexpressed Tn and STn antigens is the decreased activity of C1GalT1, which was described in some colon cancer lines [47]. This phenomenon was further uncovered by the Cummings’ lab, which showed that loss of function mutations in Cosmc, a unique chaperon for C1GalT1, causes a reduction of Core 1 extension pathway in different cancer cells, including colon, melanomaderived, and cervical cancers [48]. However, this mechanism cannot be ascribed to many other cancers, where C1GalT1 is stably expressed and localized in the fragmented Golgi [34]. This would not have been possible in cells with mutant Cosmc because dysfunction of Cosmc results in retention of C1GalT1 in the ER [49]. The key question, then, is the mechanism that underlies the overexpression of Tn and STn despite of presence of C1GalT1.
O-glycosylation is initiated by GalNAc-transferases, GalNAc-Ts, which are present throughout the Golgi stacks and preceded function of C1GalT1 (Figure 1a). Because relocation of GalNAc- Ts from Golgi to the ER has been observed in colon cancer [10] and in response to growth factor stimulation [50], it seems reasonable to assume that colon cancer-specific disassembly of Golgi is accompanied by a mistargeting of GalNAc-Ts, thus partially interrupting the initial step of O-glycosylation and reducing the sensitivity of cancer cells to TRAIL-induced apoptosis [51]. On the other hand, Golgi fragmentation may result in sub- Golgi redistribution of ST6GalNAc1 as it is described in breast cancer cells [52]. This rearrangement allows ST6GalNAc1 to successfully compete with C1GalT1 for Core 1 substrate. Similarly, the overexpression of Core 3 synthase, an enzyme that in normal mucins compete with C1GalT1 for GalNAc substrate, can be a reason for down-regulation of latter during malignancy [53,54] (Figure 1b). The Golgi mistargeting of glycosyltransferases may also be caused by the loss of their Golgi retention partner; however, despite the significant progress in our understanding of the Golgi retention mechanism of glycosyltransferases [55,56], this possibility needs further investigation.
RAS Superfamily GTPases Promote Golgi Fragmentation and Coordinate the Function of Anti-apoptotic Kinases
In the past decade, substantial progress has been made in understanding the role of Rab proteins in cancer. Overexpression or activation at least dozen of Rabs has been described in different type of cancer [57]. Among them, Rab1a [58], Rab3d [59], Rab6a [34,60,61], Rab8 [62] and Rab12 [63] are localized to different compartments of the Golgi. They tightly associate with golgins and coordinate protein transport and maintenance of a Golgi organization [64,65]. It should be noted that Rab proteins, contrary to Ras, provide cancer pathways without mutation. Furthermore, while Rac proteins are mostly monomeric [66], Rabs may form a dimer, which property increases affinity of their dimer-effectors for the Golgi membrane [67]. One powerful example is a dimeric form of Rab6a, which interacts with giantin during formation of compact Golgi. In advanced prostate cancer, however, it cooperates with Myosin IIA, providing Golgi disassembly [34] (Figure 2a). Further, the function of Rab proteins is closely associated with pathways mediated by kinases; Rab25 overexpression, for example, has been suggested to be a marker of ovarian [68] and breast cancer [69]. Notably, Rab25 was found abundantly expressed in the dispersed Golgi [70], and its overexpression increases signaling through the PI3K/Akt pathway and decreases expression of the proapoptotic BCL2 family members [71,72].
Recently the Bard’s group has found that depletion of at least 53 signaling genes induces strong fragmentation of the Golgi [73]. Among them are a wide range of kinases whose aberrant expression has been found in different types of cancer. For instance, down-regulation of inositol-trisphosphate 3-kinase A (ITPKA) was described in oral squamous cell carcinoma [74], ketohexokinase (KHK) in renal cell carcinoma [75], protein kinase D (PKD) in prostate, breast, gastric, and colon cancer [76]. The different isoforms of protein kinase C, including α, β, δ, and η is reduced in a large number of tumors, and its decrease often correlates with tumor grade [77]. However, the down-regulation of kinases during cancer development is only one side of the coin. Indeed, several pieces of evidence strongly indicate that many other kinases are upregulated; below are only a few examples.
The elevated expression of diacylglycerol kinase, zeta, and DGKζ, contributes to increased Rho GTPase activation and the enhanced motility of metastatic colorectal cancer cells [78]. Another of the Golgi-specific kinases, MAP kinase ERK8 [73,79] and the P21- activated protein kinase (Pak1) [80] are elevated in tumor cells and positively regulate cell migration. The overexpression of Src kinases results in fragmentation of Golgi in pancreatic cancer cells [81] and secretion of angiogenic growth factors [82]. Further, the members of serine/threonine protein kinases (Ste20), YSK1 and MST4 target Golgi via the golgin GM130, and their depletion alters Golgi structure and inhibits cell migration [83]. PKCε is increased in brain, bladder, and breast cancers [77], and it is detected in the Golgi upon its activation [84]. Notably, hyperactivity of kinases in cancer is accompanied by down-regulation of phosphatases, which enzymatic action is directly opposite to that of kinases. The level of dual specificity phosphatase 6 (DUSP6) is reduced in lung cancer [85], and expression of dual-specificity phosphatase 2 (DUSP2) is low in breast, colon, lung, ovary, kidney, prostate, liver, and thyroid cancer [86]. It is important to note here that knockdown of both DUSP2 and DUSP6 significantly alters Golgi morphology [73].
Thus, the kinases play a dual role during tumor progression. Perhaps the key to understanding the nature of the Janus face of kinases lies in their different response to the Golgi structure. During malignant transformation and tumor progression, the anti-apoptotic kinases are upregulated, thus facilitating survival and proliferation [76]. Their appearance in the Golgi practically coincides with Golgi disorganization, which in turn hinders Golgi targeting of proapoptotic kinases and thereby inducing their degradation. In normal cells with unaffected Golgi, these kinases negatively regulate proliferation and activate apoptosis. Therefore, the inviolability of the Golgi is an important determinant for domination of proapoptotic kinases over their anti-apoptotic counterparts and consequently for the outcome of either programmed death or survival.
Myosin Proteins and Golgi Fission
The dynamic of Golgi membranes is triggered, among other cytoskeleton proteins, by the actin cytoskeleton and by associated unconventional myosins [87]. In many cases, the upregulation of Golgi-associated myosin motors is associated with aggressive cancer. For instance, overexpression of Myosin 1b was described in head and neck squamous cell carcinoma [88], Myosin Va in colorectal cancer [89], and Myosin VI in prostate cancer [90]. The Myosin 18a directly interacts with Golgi phosphoprotein 3 (GOLPH3) and their link triggers Golgi dispersal [91]. Given that overexpressed GOLPH3 promotes proliferation and tumorigenicity [92-94], it is becoming understandable that Myosin 18 directly coordinates with Golgi morphology. It is also intriguing that GOLPH3-Myosin 18a partnership is also necessary for Golgi fragmentation induced by DNA damage, itself a prerequisite for most mutations and cancer [95].
During the past decade, increasing attention has been given to non-muscle Myosin IIA (NMIIA). The dynamic association of NMIIA with Golgi is coordinated by Rab6a, and in tandem they control retrograde transport from Golgi to ER [96]. Recent studies have shed light on the precise mechanism of NMIIA involvement in Golgi remodeling, demonstrating that NMIIA interacts with the cytoplasmic tail of Golgi glycosyltransferases, and this link provides not only the transportation of glycosyltransferases to the Golgi, but also creates a force for Golgi disorganization after: (a) heat shock or treatment with heat shock proteins inhibitors; (b) knockdown of beta-COP; and (c) treatment with Brefeldin A [13,97-99]. The ultimate role of NMIIA in cancer progression remains controversial. Some studies indicate that cessation of NMIIA results in a decrease in contractility and an increase in cell migration [100], and the level of NMIIA is diminished in human squamous cell carcinomas with poor survival [101]. Others show that the activity of NMIIA and its phosphorylation are positively correlated with the enhanced migration and invasion of tumor cells [102-104]. Our recent observations of the role of NMIIA in Golgi fragmentation also tempt us to speculate that NMIIA is a key driver of colon and prostate cancer progression. First, NMIIA is more stably associated with Golgi of androgenrefractory prostate cancer cells than androgen-sensitive cells. Second, inhibition or siRNA knockdown of NMIIA restores compact Golgi morphology in prostate and colon cancer cells [13,34]. This process is mediated through Rab6a, which loses interaction with NMIIA, thereby facilitating giantin dimerization (Figure 2a). Finally, the Golgi renaissance in advanced prostate cancer is accompanied by Golgi re-targeting of C2GnT1, which, in turn, increases susceptibility to galectin-1-induced apoptosis by replacing sialyl-T antigen with polylactosamine [34] (Figure 2b).
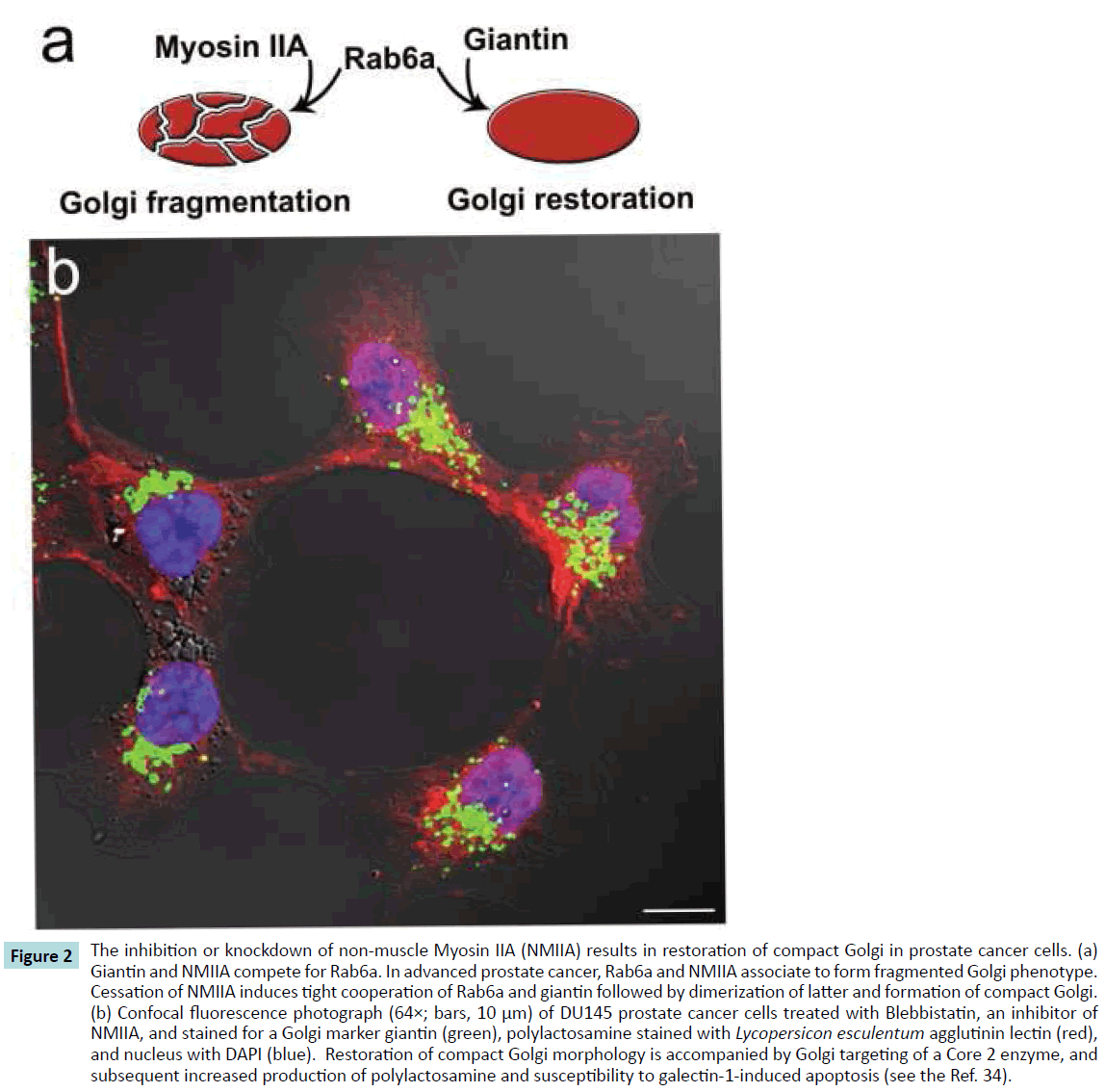
Figure 2 The inhibition or knockdown of non-muscle Myosin IIA (NMIIA) results in restoration of compact Golgi in prostate cancer cells. (a) Giantin and NMIIA compete for Rab6a. In advanced prostate cancer, Rab6a and NMIIA associate to form fragmented Golgi phenotype. Cessation of NMIIA induces tight cooperation of Rab6a and giantin followed by dimerization of latter and formation of compact Golgi. (b) Confocal fluorescence photograph (64×; bars, 10 μm) of DU145 prostate cancer cells treated with Blebbistatin, an inhibitor of NMIIA, and stained for a Golgi marker giantin (green), polylactosamine stained with Lycopersicon esculentum agglutinin lectin (red), and nucleus with DAPI (blue). Restoration of compact Golgi morphology is accompanied by Golgi targeting of a Core 2 enzyme, and subsequent increased production of polylactosamine and susceptibility to galectin-1-induced apoptosis (see the Ref. 34).
The “Tug of War” in Golgi: The Choice between Death and Survival
Fragmentation of the Golgi is an essential event in all forms of apoptosis [105]. The Golgi localized caspase-2 and caspase-3 are generally accepted as the central players in the Golgi executionphase of apoptosis, because they mediate cleavage of several golgins and GRASPs, including golgin 160 [106], giantin [107], GM130 [108], and GRASP65 [109]. The simultaneous degradation of these structural proteins results in the significant fission of Golgi. However, it is important to note that giantin is more stably associated with Golgi fragments than other golgins during apoptosis [110], confirming that giantin is the cornerstone of Golgi architecture.
What is most astonishing is the high degree of fidelity with which Golgi transforms into two daughter parts during cell division. Under normal conditions, the Golgi G2 checkpoint gives the ‘‘green light’’ for entry into mitosis [111], but when DNA is damaged cells might be stopped at the G2, thus blocking the possible development of cancer [112]. At that point, cells should undergo apoptosis. This hara-kiri mechanism induced by chemical G2 checkpoint abrogators is one of the main strategies in the modern treatment of cancer [113]. Without this mechanism, the cells become malignant and permanently exhibit G2-specific fragmented Golgi [114]. The ability of tumor cells to override apoptosis is one of the hallmarks of cancer. Several anti-apoptotic mechanisms for the suppression of proapoptotic protein are employed by cancer cells for survival, including transcriptional, translational and post-translational regulation [115]. Whether these events are accompanied by dysregulation of caspases at the Golgi is uncertain, but it would seem likely, given that caspasemediated degradation of golgins irreversibly results in apoptosis (Figure 3).
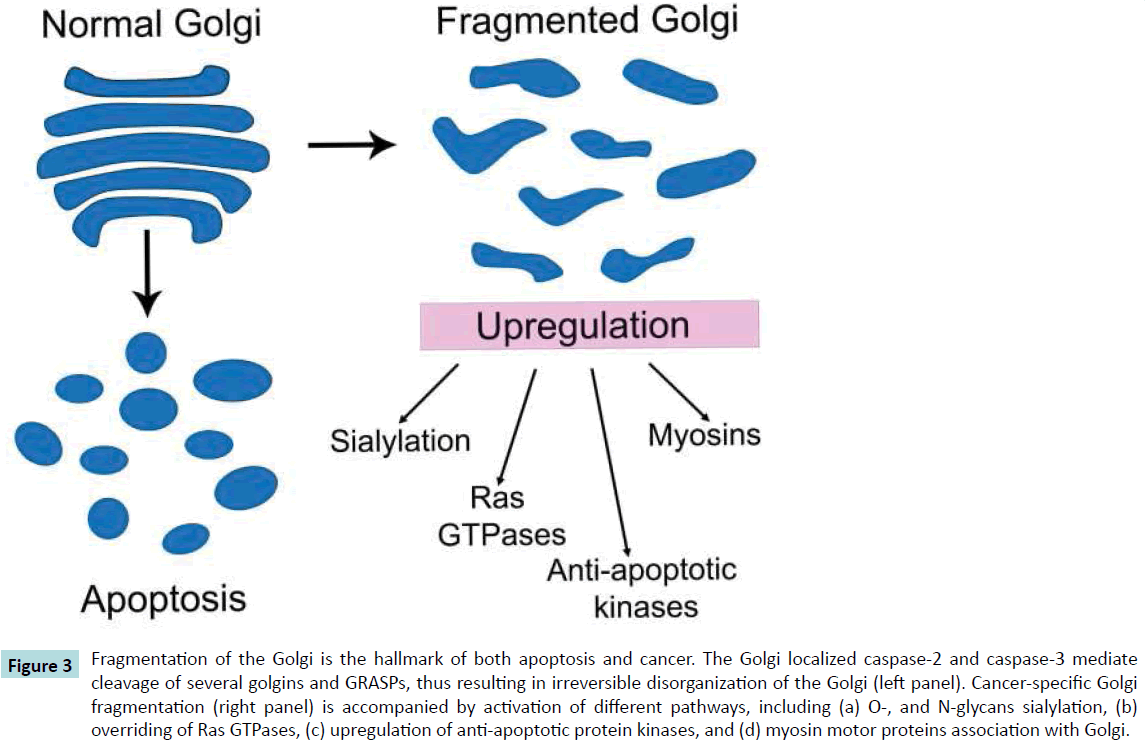
Figure 3 Fragmentation of the Golgi is the hallmark of both apoptosis and cancer. The Golgi localized caspase-2 and caspase-3 mediate cleavage of several golgins and GRASPs, thus resulting in irreversible disorganization of the Golgi (left panel). Cancer-specific Golgi fragmentation (right panel) is accompanied by activation of different pathways, including (a) O-, and N-glycans sialylation, (b) overriding of Ras GTPases, (c) upregulation of anti-apoptotic protein kinases, and (d) myosin motor proteins association with Golgi.
Targeting the Golgi as a Potential Therapeutic Intervention
Disruption of the Golgi apparatus was a promising challenge in translational research because most of these agents induce cell death. For instance, swainsonine, an inhibitor of Golgi alphamannosidase II, was considered as an anti-cancer drug with the potential for treating gastric carcinoma [116] and glioma [117]. However, a phase II clinical trial of GD0039 (a hydrochloride salt of swainsonine) in patients with renal carcinoma did not reveal any anti-tumor effect [118]. The other Golgi disruptive chemical, Brefeldin A, showed antiproliferative effects in vitro and inhibition of tumor growth in vivo [119,120], but the clinical implication has not progressed because of its poor solubility in water and its neurotoxicity. Moreover, as has been shown by BFA ester conjugates, the disruption of the Golgi complex is not necessary for cytotoxicity [121], indicating that the anti-tumor activity of BFA cannot be simply ascribed to its ability to induce Golgi collapse. The same Golgi disorganization approach was adopted in the series of preclinical studies which showed that silencing GM130 decreased angiogenesis and cells invasion in vitro and in lung cancer mice models [122]. In sum, the implication of Golgi disruptive agents looks like a dead-end, given that this strategy, in spite of its potential ability to launch apoptosis, may also accentuate Golgi fragmentation and increase the metastatic potential of cancer cells.
Over the last two decades, targeting Ras GTPAses was an attractive clinical task: at first glance, it seemed so simple to search for drugs that could interfere with GTP binding to stop mutant Ras, and in preclinical models the agents that block Ras activation through inhibition of the enzyme farnesyl transferase resulted in cell growth arrest [123]. However, in clinical studies their activity was far less promising than anticipated. Newer and more promising results came from Shokat’s lab, which found a small molecule that irreversibly binds to a common oncogenic mutant, K-Ras(G12C), but does not affect the wild-type Ras [124]. However, these compounds have also not yet passed clinical tests [125]. To date, the few encouraging results we have may give us hope to develop new Rab-specific anticancer therapies. A biphosphonate derivative, 3-PEHPC [3-(3-pyridyl)-2- hydroxy-2-phosphonopropanoic acid], inhibits posttranslational modification of Rabs, thereby inducing apoptosis of human myeloma cells in vitro [126] and reducing skeletal tumor growth in vivo [127]. It will be interesting to see what potential merits these findings deliver.
The inhibitors of NMIIA may also yield novel cancer therapies. In preclinical models, Blebbistatin have shown excellent effects on Golgi restoration [34] and the blocking of invasiveness of both MCF-7 breast cancer [102] and pancreatic adenocarcinoma cells [128]. Another important consideration are the inhibitors of S100A4, a member of the S100 family of Ca2+-binding proteins, which regulates carcinoma cell motility via interactions with NMIIA [129]. Also, great attention was paid to the possible treatment of cancer by the inhibitors of kinases. To date, several kinase inhibitors have received US Food and Drug Administration approval, but their implication is limited by mutation actions of kinases that abrogate drug binding and by their high toxicity [130].
Taken together, we believe that it is more important from a clinical perspective to target fragmented Golgi at the G2 phase, before cancer cells have passed the cell circle. The chemical abrogation of the Golgi fragmentation and its possible restoration could bring short-term control of malignancy, in which the fatal pathways described in this article will be avoided and apoptosis will be induced.
Concluding Remarks
Several important conclusions emerge from the phenomena described in this Review article. First, Golgi fragmentation results in the substantial rearrangement of Golgi residential glycosyltransferases, leading to the formation of cancer specific glycosyl epitopes. Second, Ras-proteins and myosin motor proteins are involved in the formation of disassembled Golgi phenotype. Third, the alteration of Golgi might ensure cancer cell survival by affecting the activity of proapoptotic kinases (Figure 3). It is also important to consider that downregulation of NMIIA is shown to restore compact Golgi and to increase susceptibility to galectin-1-induced apoptosis in prostate cancer cells; however, whether this phenomenon is universal and applicable to other types of cancer remains to be determined. Further studies are also needed to investigate the precise role of the GM130-GRASP65 complex and other golgins in cancer-specific remodeling of Golgi.
In sum, onco-Golgi seems the overriding condition for the survival of cancer cells. On the one hand, formation of fragmented Golgi phenotype is a cause of carcinogenesis, but on the other, it may be considered a consequence of cancer progression. Therefore, we anticipate confirmation of the existence of a vicious circle involving “Golgi fragmentation ↔ cancer progression.” The most important question is whether restoration of Golgi may block the crucial downstream pathways described in this article.
Acknowledgments
I thank Dr. Adrian E. Koesters for critical review of the manuscript. This work is supported by the K01AA022979-01 award from the National Institute on Alcohol and Alcohol Abuse (to A.P.).
References
- Stanley P (2011) Golgi glycosylation. Cold Spring HarbPerspectBiol3.
- Schultz MJ, Swindall AF, Bellis SL (2012) Regulation of the metastatic cell phenotype by sialylatedglycans. Cancer Metastasis Rev 31:501-518.
- Lee M, Lee HJ, Bae S, Lee YS (2008) Protein sialylation by sialyltransferase involves radiation resistance. Mol Cancer Res 6: 1316-1325.
- Swindall AF, Bellis SL (2011) Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J BiolChem286: 22982-22990.
- Amano M, Eriksson H, Manning JC,Detjen KM, Andre S, et al. (2012) Tumour suppressor p16(INK4a) - anoikis-favouring decrease in N/O-glycan/cell surface sialylation by down-regulation of enzymes in sialic acid biosynthesis in tandem in a pancreatic carcinoma model. FEBS J 279: 4062-4080.
- Park JJ, Yi JY, Jin YB, Lee YJ, Lee JS, et al. (2012) Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. BiochemPharmacol 83: 849-857.
- Kijima-Suda I, Miyamoto Y, Toyoshima S, Itoh M, Osawa T (1986) Inhibition of experimental pulmonary metastasis of mouse colon adenocarcinoma 26 sublines by a sialic acid: nucleoside conjugate having sialyltransferase inhibiting activity. Cancer Res 46: 858-862.
- Kuan SF, Byrd JC, Basbaum C, Kim YS (1989) Inhibition of mucin glycosylation by aryl-N-acetyl-alpha-galactosaminides in human colon cancer cells. J BiolChem 264:19271-19277.
- Bresalier RS, Niv Y, Byrd JC, Duh QY, Toribara NW, et al. (1991) Mucin production by human colonic carcinoma cells correlates with their metastatic potential in animal models of colon cancer metastasis. J Clin Invest 87:1037-1045.
- Egea G, Franci C, Gambus G, Lesuffleur T, Zweibaum A, Real FX (1993) Cis-Golgi resident proteins and O-glycans are abnormally compartmentalized in the RER of colon cancer cells. J Cell Sci 105: 819-830.
- Wang F, Goto M, Kim YS, Higashi M, Imai K, et al. (2001) Altered GalNAc-alpha-2,6-sialylation compartments for mucin-associated sialyl-Tn antigen in colorectal adenoma and adenocarcinoma. J HistochemCytochem 49: 1581-1592.
- Kellokumpu S, Sormunen R, Kellokumpu I (2002) Abnormal glycosylation and altered Golgi structure in colorectal cancer: dependence on intra-Golgi pH.FEBS Lett516: 217-224.
- Petrosyan A, Cheng PW (2013) A non-enzymatic function of Golgi glycosyltransferases: mediation of Golgi fragmentation by interaction with non-muscle myosin IIA.Glycobiology 23: 690-708.
- Springer GF (1997) Immunoreactive T and Tn epitopes in cancer diagnosis, prognosis, and immunotherapy. J MolMed 75: 594-602.
- Desai P (2000) Immunoreactive T and Tn antigens in malignancy: Role in carcinoma diagnosis, prognosis, and immunotherapy. Transfusion Med Rev 14:312-325.
- Conze T, Carvalho AS, LandegrenU, Almeida R, Reis CA, David L, et al. (2010) MUC2 mucin is a major carrier of the cancer-associated sialyl-Tn antigen in intestinal metaplasia and gastric carcinomas. Glycobiology20:199-206.
- Julien S, Videira PA, Delannoy P (2012) Sialyl-tn in cancer: (how) did we miss the target? Biomolecules 2:435-466.
- Springer GF, Desai PR, Spencer BD, Tegtmeyer H, Carlstedt SC, et al.(1995) T/Tn antigen vaccine is effective and safe in preventing recurrence of advanced breast carcinoma. Cancer Detect Prev19:374-380.
- Cipolla L, Rescigno M, Leone A,Peri F, La Ferla B, et al. (2002) Novel Tn antigen-containing neoglycopeptides: Synthesis and evaluation as anti-tumor vaccines. Bioorg Med Chem 10: 1639-1646.
- Keding SJ, Danishefsky SJ (2004) Prospects for total synthesis: A vision for a totally synthetic vaccine targeting epithelial tumors. ProcNatlAcadSci USA 101:11937-11942.
- Li Q, Anver MR, Butcher DO, Gildersleeve JC (2009) Resolving conflicting data on expression of the Tn antigen and implications for clinical trials with cancer vaccines. Mol Cancer Ther8:971-979.
- Charpin C, Pancino G, Osinaga E, Bonnier P, Lavaut MN, et al. (1992) Monoclonal antibody 83D4 immunoreactivity in human tissues: Cellular distribution and microcytophotometric analysis of immunoprecipitates on tissue sections. Anticancer Res 12: 209-224.
- Miles D, Roche H, Martin M,Perren TJ, Cameron DA, et al. (2011) Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011; 16: 1092-1100.
- Häuselmann I, Borsig L (2014) Altered tumor-cell glycosylation promotes metastasis. Front Oncol4: 1-28.
- Hung JS, Huang J, Lin YC, Huang MJ, Lee PH, et al. (2014) C1GALT1 overexpression promotes the invasive behavior of colon cancer cells through modifying O-glycosylation of FGFR2. Oncotarget5:2096-2106.
- Baldus SE, Hanisch FG, Kotlarek GM, Zirbes TK, et al. (1998) Coexpression of MUC1 mucin peptide Core and the Thomsen-Friedenreich antigen in colorectal neoplasms. Cancer 82:1019-1027.
- Singh R, Campbell BJ, Yu LG, Fernig DG, Milton JD, et al. (2001) Cell surface-expressed Thomsen-Friedenreich antigen in colon cancer is predominantly carried on high molecular weight splice variants of CD44. Glycobiology11: 587-592.
- Glinsky VV, Glinsky GV, Rittenhouse-Olson K, Huflejt ME, Glinskii OV, et al. (2001) The role of Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res 61: 4851-4857.
- Storr SJ, Royle L, Chapman CJ, Hamid UM, Robertson JF, et al. (2008) The O-linked glycosylation of secretory/shed MUC1 from an advanced breast cancer patient’s serum. Glycobiology18: 45-62.
- Ellies LG, Tsuboi S, PetryniakB, Lowe JB, Fukuda M, et al. (1998) Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity 9:881-890.
- Whitehouse C, Burchell J, Gschmeissner S, Brockhausen I, Lloyd KO, et al. (1997) A transfected sialyltransferases that is elevated in breast cancer and localizes to the medial/trans-Golgi apparatus inhibits the development of Core-2-based O-glycans. J Cell Biol 137: 1229-1241.
- Burchell J, Poulsom R, Hanby A, Whitehouse C, Cooper L, et al. (1999) An alpha2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology 9: 1307-1311.
- Dalziel M, Whitehouse C, McFarlane I, Brockhausen I, Gschmeissner S, et al. (2001) The relative activities of the C2GnT1 and ST3Gal-I glycosyltransferases determine O-glycan structure and expression of a tumor-associated epitope on MUC1. J BiolChem276:11007-11015.
- Petrosyan A, Holzapfel MS, Muirhead DE, Cheng PW (2014) Restoration of compact Golgi morphology in advanced prostate cancer enhances susceptibility to galectin-1-induced apoptosis by modifying mucin O-glycan synthesis. Mol Cancer Res 12: 1704-1716.
- Valenzuela HF, Pace KE, Cabrera PV, White R, Porvari K, et al. (2007) O-glycosylation regulates LNCaP prostate cancer cell susceptibility to apoptosis induced by galectin-1. Cancer Res 67: 6155-6162.
- Pfeffer SR (2001) Constructing a Golgi complex. J. Cell Biol155:873-875.
- Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott-Schwartz J (2001) Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol 12: 557-570.
- Barr FA, Short B (2003) Golgins in the structure and dynamics of the Golgi apparatus. CurrOpin Cell Biol 15: 405-413.
- Appenzeller-Herzog C, Hauri HP (2006) The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 119: 2173-2183.
- Petrosyan A, Ali MF, Cheng, PW (2012) Glycosyltransferase-specific Golgi targeting mechanisms. J BiolChem 287: 37621-37627.
- Koreishi M, Gniadek TJ, Yu S, Masuda J, Honjo Y, et al. (2013) The golgin tether giantin regulates the secretory pathway by controlling stack organization within Golgi apparatus. PLoS One 8:e59821.
- Asante D, Maccarthy-Morrogh L, Townley AK, Weiss MA, Katayama K, et al. (2013) A role for the Golgi matrix protein giantin in ciliogenesis through control of the localization of dynein-2. J Cell Sci126:5189-5197.
- Linstedt AD, Hauri HP (1993) Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. MolBiol Cell 4:679-693.
- Linstedt AD, Foguet M, Renz M, Seelig HP, Glick BS, et al. (1995) A C-terminally-anchored Golgi protein is inserted into the endoplasmic reticulum and then transported to the Golgi apparatus.ProcNatlAcadSci USA 92:5102-5105.
- Sönnichsen B, Lowe M, Levine T, Jämsä E, Dirac-Svejstrup B, et al. (1998) A role for giantin in docking COPI vesicles to Golgi membranes. J Cell Biol 140: 1013-1021.
- Puthenveedu MA, Bachert C, Puri S, Lanni F, Linstedt AD (2006) GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat Cell Biol8:238-248.
- Brockhausen I, Yang J, Dickinson N, Ogata S, Itzkowitz SH (1998) Enzymatic basis for sialyl-Tn expression in human colon cancer cells. Glycoconj J 15: 595-603.
- Ju T, Lanneau GS, Gautam T, Wang Y, Xia B, et al. (2008) Human tumor antigens Tn and sialylTn arise from mutations in Cosmc. Cancer Res 68: 1636-1646.
- Ju T, Aryal RP, Stowell CJ, Cummings RD (2008) Regulation of protein O-glycosylation by the endoplasmic reticulum-localized molecular chaperone Cosmc. J Cell Biol 182: 531-542.
- Gill DJ, Chia J, Senewiratne J, Bard F (2010) Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J Cell Biol 189: 843-858.
- Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, et al. (2007) Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 13:1070-1077.
- Sewell R, Bäckström M, Dalziel M, Gschmeissner S, Karlsson H, et al. (2006) The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J BiolChem281: 3586-3594.
- Brockhausen I (1999) Pathways of O-glycan biosynthesis in cancer cells. BiochimBiophysActa 1473: 67-95.
- Robbe-Masselot C, Herrmann A, Maes E, Carlstedt I, Michalski JC, et al. (2009) Expression of a core 3 disialyl-Le(x) hexasaccharide in human colorectal cancers: a potential marker of malignant transformation in colon. J Proteome Res 8:702-711.
- Ali MF, Chachadi VB, Petrosyan A, Cheng PW (2012) Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling Golgi localization of Core 2 N-acetylglucosaminyltransferase 1. J BiolChem 287: 39564-39577.
- Petrosyan A, Ali MF, Cheng PW (2015) Keratin 1 plays a critical role in golgi localization of Core 2 N-acetylglucosaminyltransferase M via interaction with its cytoplasmic tail. J BiolChem290: 6256-6269.
- Goldenring JR (2013) Acentral role for vesicle trafficking in epithelial neoplasia: intracellular highways to carcinogenesis. Nat Rev Cancer 13: 813-820.
- Shimada K, Uzawa K, Kato M, Endo Y, Shiiba M, et al. (2005) Aberrant expression of RAB1A in human tongue cancer. Br J Cancer 92: 1915-1921.
- Yang J, Liu W, Lu X, Fu Y, Li L, et al. (2015) High expression of small GTPase Rab3D promotes cancer progression and metastasis. Oncotarget 6: 11125-11138.
- Hennigan RF, Moon CA, Parysek LM, Monk KR, Morfini G, et al. (2013) The NF2 tumor suppressor regulates microtubule-based vesicle trafficking via a novel Rac, MLK and p38(SAPK) pathway. Oncogene 32: 1135-1143.
- Huang H, Jiang Y, Wang Y, Chen T, Yang L, et al. (2015) miR-5100 promotes tumor growth in lung cancer by targeting Rab6. Cancer Lett 362: 15-24.
- Bravo-Cordero JJ, Marrero-Diaz R, Megias D, Genis L, Garcia-Grande A, et al. (2007) MT1-MMP proinvasive activity is regulated by a novel Rab8-dependentexocytic pathway. EMBO J 26: 1499-1510.
- Matsui T, Fukuda M (2013) Rab12 regulates mTORC1 activity and autophagy through controlling the degradation of amino-acid transporter PAT4. EMBO Rep 14: 450-457.
- Sinka R, Gillingham AK, Kondylis V, Munro S (2008) Golgi coiled-coil proteins contain multiple binding sites for Rab family G proteins. J Cell Biol183:607-615.
- Liu S, Storrie B (2012) Are Rab proteins the link between Golgi organization and membrane trafficking? Cell Mol Life Sci69:4093-4106.
- Oxford G, Theodorescu D (2003) Ras superfamily monomeric G proteins in carcinoma cell motility. Cancer Lett189: 117-128.
- Pylypenko O, Attanda W, Gauquelin C, Lahmani M, Coulibaly D, et al. Structural basis of myosin V RabGTPase-dependent cargo recognition.ProcNatlAcadSci USA 110: 20443-20448.
- Davidson B, Zhang Z, Kleinberg L, Li M, Florenes VA, et al. (2006) Gene expression signatures differentiate ovarian/peritoneal serous carcinoma from diffuse malignant peritoneal mesothelioma. Clin Cancer Res 12: 5944-5950.
- Yin YX, Shen F, Pei H, Ding Y, Zhao H, et al. (2012) Increased expression of Rab25 in breast cancer correlates with lymphatic metastasis. TumourBiol 33: 1581-1587.
- Agarwal R,Jurisica I, Mills GB, Cheng KW (2009) The emerging role of the RAB25 small GTPase in cancer. Traffic 10: 1561-1568.
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, et al. (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev 11: 701-713.
- Cheng KW, Lahad JP, Kuo WL,Lapuk A, Yamada K, et al. (2004) The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat Med 10:1251-1256.
- Chia J, Goh G, Racine V, Ng S, Kumar P, et al. (2012) RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. MolSystBiol8: 629.
- Kato H, Uzawa K, Onda T, Kato Y, Saito K, et al. (2006) Down-regulation of 1D-myo-inositol 1,4,5-trisphosphate 3-kinase A protein expression in oral squamous cell carcinoma. Int J Oncol 28: 873-881.
- Hwa JS, Kim HJ, Goo BM, Park HJ, Kim CW, et al. (2006)The expression of ketohexokinase is diminished in human clear cell type of renal cell carcinoma. Proteomics 6:1077-1084.
- Sundram V, Chauhan SC, Jaggi M (2011) Emerging roles of protein kinase D1 in cancer. Mol Cancer Res 9:985-996.
- Griner EM, Kazanietz MG (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 7: 281-294.
- Cai K, Mulatz K, Ard R, Nguyen T, Gee SH (2014) Increased diacylglycerol kinase ζ expression in human metastatic colon cancer cells augments Rho GTPase activity and contributes to enhanced invasion. BMC Cancer 14: 208.
- Chia J, Tham KM, Gill DJ, Bard-Chapeau EA, Bard FA (2014) ERK8 is a negative regulator of O-GalNAc glycosylation and cell migration. Elife 3: e01828
- Ching YP, Leong VY, Lee MF, Xu HT, Jin DY, et al. (2007) P21-activated protein kinase is overexpressed in hepatocellular carcinoma and enhances cancer metastasis involving c-Jun NH2-terminal kinase activation and paxillinphosphorylation. Cancer Res 67: 3601-3608.
- Weller SG, Capitani M, Cao H, Micaroni M,Luini A, et al. (2010) Src kinase regulates the integrity and function of the Golgi apparatus via activation of dynamin 2. ProcNatlAcadSci USA 107: 5863-5868.
- Kim LC, Song L, Haura EB (2009) Src kinases as therapeutic targets for cancer. Nat Rev ClinOncol6:587-595.
- Preisinger C, Short B, De Corte V, Bruyneel E, Haas A, et al. (2004) YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol 164: 1009-1020.
- Lehel C, Olah Z, Jakab G, Anderson WB (1995) Protein kinase C epsilon is localized to the Golgi via its zinc-finger domain and modulates Golgi function. ProcNatlAcadSci USA 92: 1406-1410.
- Diaz-Garcia CV, Agudo-Lopez A, Perez C, Prieto-Garcia E, Iglesias L, et al. (2015) Prognostic value of dual-specificity phosphatase 6 expression in non-small cell lung cancer. TumourBiol36:1199-1206.
- Lin SC, Chien CW, Lee JC, Yeh YC, Hsu KF, et al. (2011) Suppression of dual-specificity phosphatase-2 by hypoxia increases chemoresistance and malignancy in human cancer cells. J Clin Invest 121:1905-1916.
- Allan VJ, Thompson HM, McNiven MA (2002) Motoring around the Golgi. Nat Cell Biol 4: E236-E242.
- Ohmura G, Tsujikawa T, Yaguchi T, Kawamura N, Mikami S, et al. (2015) Aberrant Myosin 1b Expression Promotes Cell Migration and Lymph Node Metastasis of HNSCC. Mol Cancer Res 13: 721-731.
- Lan L, Han H, Zuo H, Chen Z, Du Y, et al. (2010) Upregulation of myosin Va by Snail is involved in cancer cell migration and metastasis. Int J Cancer 126:53-64.
- Dunn TA, Chen S, Faith DA, Hicks JL, Platz EA, et al. (2006) A novel role of myosin VI in human prostate cancer. Am J Pathol 169: 1843-1854.
- Dippold HC, Ng MM, Farber-Katz SE, Lee SK, Kerr ML, et al. (2009) GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139:337-351.
- Hua X, Yu L, Pan W, Huang X, Liao Z, et al. Increased expression of Golgi phosphoprotein-3 is associated with tumor aggressiveness and poor prognosis of prostate cancer. Diagn. Pathol 7: 127.
- Zeng Z, Lin H, Zhao X, Liu G, Wang X, et al. (2012) Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin Cancer Res 18: 4059-4069.
- Hu BS, Hu H, Zhu CY, Gu YL, Li JP (2013) Overexpression of GOLPH3 is associated with poor clinical outcome in gastric cancer. TumourBiol 34: 515-520.
- Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing M, et al. (2014) DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 156: 413-427.
- Miserey-Lenkei S, Chalancon G, Bardin S, Formstecher E, Goud B, et al. (2010) Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex. Nat Cell Biol 12: 645-654.
- Duran JM, Valderrama F, Castel S, Magdalena J, Tomas M, et al. (2003) Myosin motors and not actin comets are mediators of the actin-based Golgi-to-endoplasmic reticulum protein transport. MolBiol Cell 14:445-459.
- Petrosyan A, Ali MF, Verma SK, Cheng H, Cheng PW (2012) Non-muscle myosin IIA transports a Golgi glycosyltransferase to the endoplasmic reticulum by binding to its cytoplasmic tail. Int J Biochem Cell Biol 44: 1153-1165.
- Petrosyan A, Cheng PW (2014) Golgi fragmentation induced by heat shock or inhibition of heat shock proteins is mediated by non-muscle myosin IIA via its interaction with glycosyltransferases. Cell Stress Chaperones 19: 241-254.
- Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, et al. (2007) Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol9: 299-309.
- Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, et al. (2014)Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science 343:309-313.
- Derycke L, Stove C, Vercoutter-Edouart AS, De Wever O, Dolle L, et al. (2011) The role of non-muscle myosin IIA in aggregation and invasion of human MCF-7 breast cancer cells. Int J DevBiol55:835-540.
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol10:778-790.
- Betapudi V, Licate LS, Egelhoff TT (2006) Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res 66: 4725-4733.
- Machamer CE (2003) Golgi disassembly in apoptosis: cause or effect? Trends Cell Biol13:279-281.
- Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, et al. (2000) Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol 149: 603-612.
- Lowe M, Lane JD, Woodman PG, Allan VJ (2004)Caspase-mediated cleavage of syntaxin 5 and giantin accompanies inhibition of secretory traffic during apoptosis. J Cell Sci 117: 1139-1150.
- Walker A, Ward C, Sheldrake TA, Dransfield I, Rossi AG, et al. (2004) Golgi fragmentation during Fas-mediated apoptosis is associated with the rapid loss of GM130. BiochemBiophys Res Commun316: 6-11.
- Lane JD, Lucocq J, Pryde J, Barr FA, Woodman PG, et al. (2002) Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J Cell Biol 156: 495-509.
- Nozawa K (2004) Giantin is the major Golgi autoantigen in human anti-Golgi complex sera. Arthritis Res Ther6:R95-R102.
- Sütterlin C, Hsu P, Mallabiabarrena A, Malhotra V (2002) Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 109:359-369.
- Stark GR, Taylor WR (2004) Analyzing the G2/M checkpoint. Methods MolBiol 280: 51-82.
- Kawabe T (2004) G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther3:513-519.
- Corda D, Barretta ML, Cervigni RI, Colanzi A (2012) Golgi complex fragmentation in G2/M transition: An organelle-based cell-cycle checkpoint. IUBMB Life 64:661-670.
- Fernald K, Kurokawa M (2013) Evading apoptosis in cancer. Trends Cell Biol 23: 620-633.
- Sun JY, Zhu MZ, Wang SW, Miao S, Xie YH, et al. (2007) Inhibition of the growth of human gastric carcinoma in vivo and in vitro by swainsonine. Phytomedicine14:353-359.
- Sun JY, Yang H, Miao S, Li JP, Wang SW, et al. (2009) Suppressive effects of swainsonine on C6 glioma cell in vitro and in vivo. Phytomedicine16: 1070-1074.
- Shaheen PE, Stadler W, Elson P, Knox J, Winquist E, et al. (2005) Phase II study of the efficacy and safety of oral GD0039 in patients with locally advanced or metastatic renal cell carcinoma. Invest. New Drugs 23: 577-581.
- Sausville EA, Duncan KL, Senderowicz A, Plowman J, Randazzo PA, et al. (1996) Antiproliferative effect in vitro and antitumor activity in vivo of brefeldin A. Cancer J Sci Am 2: 52-58.
- Rajamahanty S, Alonzo C, Aynehchi S, Choudhury M, Konno S (2010) Growth inhibition of androgen-responsive prostate cancer cells with brefeldin A targeting cell cycle and androgen receptor. J Biomed Sci17: 5.
- Anadu NO, Davisson VJ, Cushman M (2006) Synthesis and anticancer activity of brefeldinA ester derivatives. J Med Chem 49: 3897-3905.
- Chang SH, Hong SH, Jiang HL, Minai-Tehrani A, Yu KN, et al. GOLGA2/GM130, cis-Golgi matrix protein, is a novel target of anticancer gene therapy. MolTher 20: 2052-2063.
- Appels NM, Beijnen JH, Schellens JH (2005) Development of farnesyltransferase inhibitors: a review. Oncologist 10: 565-578.
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503: 548-551.
- Ledford H (2015) Cancer: The Ras renaissance. Nature 520: 278-280.
- Roelofs AJ, Hulley PA, Meijer A, Ebetino FH, Russell RG, et al. (2006) Selective inhibition of Rabprenylation by a phosphonocarboxylate analogue of risedronate induces apoptosis, but not S-phase arrest, in human myeloma cells. Int J Cancer 119: 1254-1261.
- Fournier PG, Daubine F, Lundy MW, Rogers MJ, Ebetino FH, et al. (2008) Lowering bone mineral affinity of bisphosphonates as a therapeutic strategy to optimize skeletal tumor growth inhibition in vivo. Cancer Res 68: 8945-8953.
- Duxbury MS, Ashley SW, Whang EE (2004) Inhibition of pancreatic adenocarcinoma cellular invasiveness by blebbistatin: a novel myosin II inhibitor. BiochemBiophys Res Commun313:992-997.
- Malashkevich VN, Dulyaninova NG, Ramagopal UA, Liriano MA, Varney KM, et al. Phenothiazines inhibit S100A4 function by inducing protein oligomerization. ProcNatlAcadSci USA 107: 8605-8610.
- Zhang J, Yang PL, Gray NS (2009) Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9: 28-39.

