Keywords
Biological Therapy; gemcitabine; Gene Expression; Pancreatic Neoplasms; Pharmacogenetics
Abbreviations
CDA: cytidine deaminase; dCK: deoxycytidine kinase; dFdCDP: difluorodeoxycytidine diphosphate; dFdCDP: ifluorodeoxycytidine 5'- diphosphate; dFdCTP: difluorodeoxycytidine 5'-triphosphate; dFdU: 2',2'-difluorodeoxyuridine; EMAP II: Endothelial Monocyte Activating Polypeptide II; (ERCC: excision repair cross complementing; ESA: epithelial surface antigen; EXO1: exonuclease 1; FAK: focal adhesion kinase; FGF: fibroblast growth factor; GAGE1: G antigen 1; MLH: mutL homolog; MMR: mismatch repair; MSH: mutS homolog; NCS: neoadjuvant candidate score; PDGF: plateletderived growth factor; PI3K: phosphoinositide-3-kinase; PMS1: postmeiotic segregation increased 1; RTOG: Radiation Therapy Oncology Group; SNPs: single nucleotide polymorphisms; TKI: tyrosine kinase inhibitor; TKI258: 4-amino-5-fluoro-3-(5-(4- methylpiperazin-1-yl)-1H-benzimidazol-2-yl)quinolin-2(1H)-one; TP73: tumor protein p73; TREX1: three prime repair exonuclease 1; XRCC: X-ray repair cross complementing
Introduction
Pancreatic cancer is one of the most aggressive and deadly malignancies with incidence equal to mortality with 37,680 estimated new cases and 34,290 deaths in 2008 only in United States [1]. It is currently the tenth commonest cancer and the fourth leading cause of cancer death in Western countries. The significant clinical and preclinical research over the last decades has not led to measurable benefits to our patients and currently strong evidence exists only for two chemotherapy drugs (gemcitabine and capecitabine), one targeted agent (erlotinib) and possibly for radiotherapy in locally advanced disease [2]. The grim prognosis of this disease along with increasing understanding of the genetics, pathogenesis and molecular dysregulations along with advances in diagnostic means allows and justifies continuation of further research.
Translational Clinical Studies
I. Pharmacogenetics Related to Gemcitabine
Gemcitabine is a fluorine-substituted deoxycytidine analog. Gemcitabine is activated intracellularly to a monophosphate form by the enzyme deoxycytidine kinase (dCK) and metabolised then to the cytotoxic nucleotide difluorodeoxycytidine 5'-triphosphate (dFdCTP). The dFdCTP metabolite is able to inhibit the actions of several DNA polymerases interfering thus to DNA chain elongation, synthesis or DNA repair. The diphosphate form (dFdCDP) inhibits the enzyme ribonucleotide reductase, resulting in decreased levels of essential deoxyribonucleotides for DNA synthesis and function (Figure 1).
Figure 1. Metabolism of gemcitabine. Adapted and modified from
Saif MW [12].
Incorporation of dFdCTP into RNA results similarly in alterations in RNA processing and mRNA translation. Gemcitabine is metabolized to its inactive metabolite, 2',2'-difluorodeoxyuridine (dFdU) by cytidine deaminase (CDA). CDA is a polymorphic enzyme and there is one variant allele (208G>A (3*, A70T)) that was identified in Japanese population (allele frequency of 0.037) that had functional impact of CDA. 3* allele appeared to change pharmacokinetic parameters and plasma CDA activities significantly leading to decreased clearance of gemcitabine and increased toxicity [3]. In this cohort of Japanese patients the variant 79A>C (2*, Lys27Gln) with frequency of 0.207 was also studied and no significant effect on gemcitabine pharmacokinetics was reported. More polymorphisms have been identified and their functional impact will need to be examined [4, 5].
Farrell et al. studied the prognostic and predictive value of the aforementioned single nucleotide polymorphism (SNP) (79A>C(Lys27Gln)) of CDA in patients treated with adjuvant treatment in the phase III study of the Radiation Therapy Oncology Group (RTOG 9704), which compared radiotherapy plus 5- FU to radiotherapy plus gemcitabine (Abstract #115). This CDA genotypic SNP was tested separately in both treatment arms in association to overall survival, disease free survival, and toxicity. Eighty-seven patients were eligible for analysis and the authors reported increasing hematological toxicity in patients with the homozygous reference (A/A, Lys/Lys) genotype compared to variant (C/C, Gln/Gln) (P<0.001) as well as compared to heterozygous genotype (A/C, Lys/Gln) (P<0.03). This difference in hematological toxicity was not observed in the 5-FU arm. Genotype variations were not associated with differences in overall survival and disease free survival. The authors concluded that the nonsynonymous CDA (79A>C (Lys27Gln)) SNP is confirmed as a predictive marker of gemcitabine hematologic toxicity, but not prognostic of treatment response.
The contradictory result compared to the study in Japanese patients may have to do with ethnic and racial variations as the RTOG 9704 study was performed on Australian and North American patients.
Upon damage, DNA strands are repaired by various enzymes predestinated for this purpose such as ERCC (excision repair cross complementing), XRCC (X-ray repair cross complementing) and mismatch repair (MMR) enzymes. Genes encoding for these proteins (ERCC, XRCC, mutL homolog 1 (MLH1), mutS homolog 2 (MSH2), MSH6, etc.) are subject to SNPs altering their functions and their activity.
Researchers from M.D. Anderson Cancer Center, United States, evaluated 15 SNPs of eight MMR genes (exonuclease 1 (EXO1), MLH1, MSH2, MSH3, MSH6, postmeiotic segregation increased 1 (PMS1), three prime repair exonuclease 1 (TREX1), and tumor protein p73 (TP73)) in 154 patients with potentially resectable pancreatic adenocarcinoma enrolled in phase II studies of gemcitabine based preoperative chemoradiotherapy (Abstract #118). They subsequently associated these genotypes to treatment response, clear surgical margins and overall survival. They concluded that three genotypes (five, six and ten) were associated significantly with tumor response, respectability and overall survival in univariate analysis. Favorable genotypes for tumor response included TP73 GG (versus the GA/AA carriers), TREX1 EX14-460C>T and TP73 Ex2+4G>A. For tumor respectability the favorable SNPs included TP73 Ex2+4G>A GG (compared to GA/AA carriers) and genotypes MLH1 IVS12- 169C>T and TP73. The genotypes EXO1 R354H, TREX1 and TP73 were significant predictors of overall survival in multivariable models including all clinical factors. An additive genotypic effect on these clinical endpoints was observed, i.e. patients with 0-1 adverse genotypes are still alive and those with more adverse genotypes demonstrated a decreasing median survival from 36.2 months (2 adverse SNPs) to 8.3 months (6-7 adverse SNPs) (P<0.001). Therefore, it seems that polymorphisms of MMR genes may potentially serve as predictors of treatment response to gemcitabine-based therapy and as prognostic factors for tumor resection and overall survival of patients with localized disease.
As pancreatic cancer demonstrates a dismal prognosis and response of the total population to treatment is moderate at best, pharmacogenomics play an increasing role in patients’ selection, in order to maximize efficacy and minimize unnecessary toxicities. In a study on fresh frozen samples from resected pancreatic cancers, RNA analysis and gene expression profile was undertaken (Abstract #142). The researchers used the median survival of the patients (13 months, range 2-53 months) as the time point in order to classify the samples into two groups. Differential gene expression analysis (fold change greater than 2, difference of means greater than 100; P<0.05) revealed 21 probe sets. They performed hierarchical clustering of the samples, using these 21 probe sets, and displayed two separate cluster sets. One cluster contained only samples from patients with a survival time less than 13 months. Based on the cluster data, this method demonstrated a 100% sensitivity and 73% specificity for the detection of samples from patient with a survival greater than 13 month.
II. EGFR Pathway Related Mutations
The epidermal growth factor receptor (EGFR) -> RAS -> PI3K (phosphoinositide-3-kinase) pathway has been found overactivated in various solid cancers including pancreatic cancer. Mutations leading to constitutive amplification of the involved molecules are not uncommon and are often related to drug resistance or drug sensitivity (e.g., resistance to EGFR inhibitors in K-ras mutations in colon cancer and susceptibility to EGFR inhibitor gefitinib in EGFR gene mutations in non-small cell lung cancer). In Abstract #173, Iver et al. studied the prevalence of PI3K catalytic subunit (PI3KCA) and EGFR mutations in tumors from 30 pancreatic cancer patients who underwent Whipple’s procedure and found no convincing evidence of association and therefore no role of screening for these mutations in pancreatic cancer
Clinical observation and physical examination remain still unsurpassed tools of treatment response assessment, even at the era of the very sophisticated therapies and technologies. One of the proposed clinical biomarkers of treatment with EGFR monoclonal antibodies or small molecules is the development of skin rash (folliculitis), the underlying mechanism of which seems to be likely immunological though not fully explored.
III. Rash as a Surrogate Marker of Response
Van Cutsem et al. presented the results from the phase III AViTA study (gemcitabine + erlotinib ± bevacizumab), with reference to skin rash as a marker of efficacy of gemcitabine plus erlotinib based therapy in pancreatic cancer (Abstract #117). They reported that higher grade of rash was associated with higher overall survival in both treatment arms; though no statistical difference in overall survival was observed between the two arms for rash of any grade (Table 1). These results were similar to the findings from the previous phase III PA.3 study by Moore et al. (gemcitabine±erlotinib) [6].

IV. Predictive Factors of Early Mortality Following Palliative Bypass
Surgery remains the definitive curative treatment for the minority of pancreatic cancer patients who present at an early stage. Despite the mounting surgical experience and treatment at specialist tertiary cancer centers the majority of the patients undergoing surgery will relapse and succumb to their disease. Quite often patients are found to be inoperable only at the time of surgery and therefore may undergo a palliative procedure. Thus, there is a need for a more accurate selection of patients considered for surgery who may though benefit from a preoperative strategy.
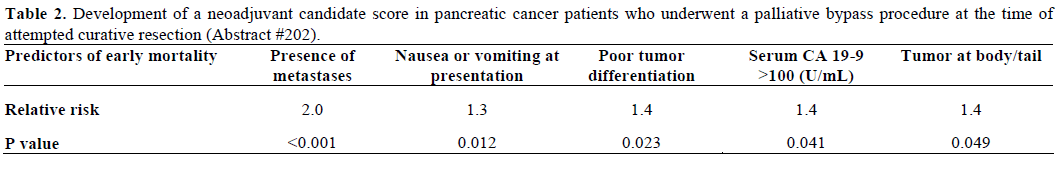
Gray et al. conducted a retrospective study aiming to identify the predictive factors of early mortality following palliative bypass (during an attempt curative surgery). They also tried to develop a candidate score to predict the group of patients who may benefit from neoadjuvant treatment. They evaluated 402 patients and the predictive factors of early mortality (less than 6 months) in univariate analysis are illustrated in Table 2. Interestingly, age, race, sex, tumor size, preoperative weight loss and comorbidities were not associated with early mortality. They researchers found five statistically significant predictors each of which was assigned one point and a neoadjuvant candidate score (NCS) was created. Patients with NCS 0 were more likely to survive beyond six months compared to patients with NCS 1 (P=0.001) and NCS greater than 1 (P<0.0001). Therefore, this information may be useful in identifying patients who may benefit from upfront preoperative treatment.
V. Cancer Stem Cells
It has been suggested previously in various solid cancers and hematological malignancies that carcinogenesis and cancer progression depends partly on the presence of cancer stem cells which via selfrenewal and differentiation properties are able to drive tumorigenesis and resist to conventional anticancer therapies [7, 8, 9]. These cancer stem cells often express distinct cell surface antigens which we are able to detect and identify the population. The main cell surface antigens recognized in cancer stem cells in various malignancies include CD44+, CD24+/-, epithelial surface antigen (ESA)+ and CD133+ [8, 10, 11]. In pancreatic cancer the known cancer stem cell markers so far are CD44+, CD24+ and ESA+.
Kim et al. studied the presence and expression of the surface antigen CD133+ in pancreatic cancer mouse xenografts derived directly from pancreatic cancer patients (Abstract #150). Four mouse xenografts from three patients were screened for CD133+ cells. One xenograft was derived from a patient previously treated with preoperative chemotherapy, and two xenografts from the same patient (one from his primary and the other from his lymph node metastasis). They found that CD133+ cells were observed in all xenografts (mean cell population was 13.5%, range 1.5-25.3%). The CD133+ populations were almost the same in xenografts derived from the primary tumor of two patients, regardless of preoperative treatment status (13.4% and 13.8%). In xenografts derived from the third patient, observed CD133+ cell populations in primary tumor and lymph nodes metastases were 1.5% and 25.3%, respectively. Though the study sample size was small, the authors suggested that CD133+ marker is useful in identifying pancreatic cancer stem cells both from primary and metastatic sites, regardless the treatment status.
The disappointing results of the current treatments in pancreatic cancer have called for research of other pathways of targeting the tumors and their pathophysiological mechanisms. One of the under evaluation, in clinical studies, concepts is immunotherapy which involves the development of antibodies, peptides, vaccines and recombinant viral and/or bacterial vectors targeting tumor antigens or inducing B- or T-cells responses able to cause tumor regression and rejection. It is known that pancreatic cancers often overexpress genes of the G antigen (GAGE) family (especially GAGE1) which encode for tumor-specific antigens presented by HLA I molecules and which are recognized on tumor cells by cytotoxic T lymphocytes. Therefore, these genes may serve as targets of immunotherapy
VI. GAGE1 Gene
Batchu et al. (Abstract #178) confirmed the expression of GAGE1 gene in various pancreatic cancer lines. They also reported the successful development of a recombinant adeno-associated viral (rAAV) vector carrying GAGE1 gene able for B-cells transduction and therefore potentially able to induce cytotoxic T lymphocyte (CTL) responses.
VII. Targeting Focal Adhesion Kinase (FAK)
Remaining at the field of targeted therapies, Hochwald et al (Abstract #144) reported the development of a FAK (a cytoplasmic tyrosine kinase) small molecule inhibitor, called 1,2,4,5-benzenetetraamine tetrahydrochloride (or Y drug). Y drug principal action is the targeting of the main autophosphorylation site of FAK. Its effect on human pancreatic cancer cell biochemistry, cell viability, adhesion, apoptosis and tumor growth in vivo was tested (Miapaca-2, Panc-1 cells were used). In this study, Y drug blocked phosphorylation of FAK at doses of 1-100 μM. The direct inhibition of FAK autophosphorylation was dose-dependent. Other significant dose-dependent effects of Y drug included increased cell detachment and inhibition of cell adhesion and viability (P<0.05). Even at the low starting dose of 5 μM, Y drug caused poly(ADP-ribose) polymerase and caspase-3 cleavage in pancreatic cancer cells indicating apoptosis and anticancer activity. In vivo, Y drug (30 mg/kg via i.p. injection daily) effectively and significantly caused pancreatic tumor regression. This effect was enhanced, when administered along with i.p. gemcitabine (30 mg/kg) chemotherapy. The inhibition of FAK was related to increasing apoptosis (as evidenced by increase of the caspase-3 cleavage) and decreasing tumor proliferation (as evidenced by decrease of the Ki67 proliferation index). The authors concluded that targeting the Y397 autophosphorylation site of FAK in pancreatic cancer with the small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride (Y drug), deserves further investigation as a novel treatment strategy in pancreatic cancer.
VIII. Targeting Vascular Endothelial Growth Factor Receptor (VEGFR)
Another novel agent TKI258 (4-amino-5-fluoro-3-(5- (4-methylpiperazin-1-yl)-1H-benzimidazol-2- yl)quinolin-2(1H)-one; small molecule, multiple receptor kinase inhibitor) was tested in a preclinical study and presented by Lang et al. (Abstract #145). TKI258 inhibits receptors to vascular endothelial growth factor (VEGFR), platelet-derived growth factor (PDGFR) and basic fibroblast growth factor (bFGFR) all of which are involved in formation of metastases and angiogenesis affecting pancreatic cancer cells, endothelial cells) and vascular smooth muscle cells. The researchers reported that TKI258 impaired pancreatic cancer cell, endothelial cell and vascular smooth muscle cell growth in a dose-dependent fashion. Furthermore, TKI258 affected tumor cell migration induced by bFGF and FGF-7 (P<0.05). In pancreatic cancer cells treatment with TKI258 resulted in reductions of N-cadherin and survivin expression. Similarly, in endothelial cells this TKI caused reduction of the constitutive and VEGF-A-mediated cell motility (P<0.05), inhibition of VEGFR and of the downstream effector FAK activation and decrease of delta-like 4 (DLL4) mRNA expression. In vascular smooth muscle cells, TKI258 treatment caused impairment of the PDGF-B-induced activation of signaling intermediates. The above results provide evidence, according to the investigators, that inhibition of multiple receptor tyrosine kinases by TKI258 could be valuable for anti-angiogenic and anti-metastatic therapy of pancreatic cancer. As such, more in vivo studies of TKI258 effect on pancreatic cancer are currently in progress.
IX. Endothelial Monocyte Activating Polypeptide II (EMAP II)
A further interesting study tested the role of maintenance of anti-endothelial combination therapy on long-term survival in experimental pancreatic cancer (Abstract #158). Schwarz et al. evaluated the benefits of EMAP II, an antiangiogenic, antiendothelial cytokine, in combination treatment of localized pancreatic adenocarcinoma. They used gemcitabine-resistant human pancreatic adenocarcinoma cells AsPC in murine xenograft. The models were treated with various combinations of EMAP II (E, 80 μg/kg i.p. daily), bevacizumab (B) 2.5 mg/kg i.p. twice weekly), or gemcitabine (G, 100 mg/kg i.p. twice weekly) for 14 days, or continued until death (n=8 each group). They also tested the in vitro the combination treatment effects on AsPC and human umbilical vein endothelial cells. According to the authors EMAP II contributed significantly to the cells survival. The median survival was 23 days in controls, 32 days in the B+G group, 42 days in the E+B+G group, and 48 days in continued E+B+G (P<0.0001). The added survival benefit of EMAP II to B+G was statistically significant in all groups. Treatment of AsPC cells with EMAP II dose up to 20 μM alone or in combination with B+G did not show any antiproliferative effect. On the other hand, growth of human umbilical vein endothelial cells endothelial cells was inhibited with EMAP II, an effect that was observed enhanced in combination with B+G. Finally pre-incubation of human umbilical vein endothelial cells with 20 μM EMAP II was found to inhibit: a) binding of VEGF to its receptors R1 and R2; b) VEGFmediated receptor phosphorylation; and c) activation of downstream VEGF signaling molecules such as v-akt murine thymoma viral oncogene homolog (AKT), elkrelated tyrosine kinase (ERK), p38 and raf. Therefore, subject to validation of these results in other studies, EMAP II may contribute in future in the management of pancreatic cancer.
X. Pretargeted Radio-Immunotherapy
The role of pretargeted radio-immunotherapy of pancreatic cancer xenografts in nude mice with a humanized, recombinant bispecific antibody (bsMAb) and a 90Y-labeled hapten-peptide was explored in Abstract #163. In this study, mice bearing human pancreatic tumor, were injected with a recombinant bsMAb, TF10, followed one day later by a 90Y-labeled hapten-peptide (IMP-288). Gemcitabine chemotherapy was added in various doses and schedules. Tumor progression was monitored for up to 28 weeks. Pretargeted radio-immunotherapy (0.7 mCi) alone caused a transient 60% reduction in blood counts. Mice treated with 0.9 mCi of pretargeted radioimmunotherapy alone and 0.7 mCi pretargeted radioimmunotherapy with 6 mg gemcitabine (equivalent to 1,000 mg/m2 in humans) had no histological evidence of renal toxicity after 9 months. Single dose of 0.25 or 0.5 mCi pretargeted radio-immunotherapy alone resulted in 20% to 80% regression of the tumors, respectively. Monthly fractions of pretargeted radioimmunotherapy (0.25 mCi/dose at the start of each gemcitabine cycle) along with standard gemcitabine treatment (6 mg weekly x 3; 1 week off; for 3 cycles) significantly increased the median time for tumors to reach 3.0 cm3 over pretargeted radio-immunotherapy alone. In conclusion, pretargeted radio-immunotherapy may enhance therapeutic responses in pancreatic cancer treatment either alone or by acting as a chemosensitizer.
Discussion
There is no doubt there is a currently florid research activity in the majority of solid tumors including pancreatic cancer. Increasing research in pancreatic cancer is mainly driven by the rising incidence in a gradually ageing population and the still incomplete understanding of the mechanisms of its carcinogenesis. This realit is projected to current empirical and nonetiological therapies. The pace of disease progression in pancreatic cancer is exceptionally rapid. Due to poor prognosis there is no enough time to use various old and novel agents alone, together or in various sequences in order to improve our results. Hence, international collaboration and integration of translational and clinical research is more than ever needed and applicable.
In conclusion, we should congratulate the researchers for their determination anticipating with particular interest the results of the next stage of development of each of the above findings, hoping for more sophisticated methods and treatments. At the same time, we should bear in mind the “bottle-neck” phenomenon observed in drug development and in novel applications which eventually reach and benefit the individual patient.
Conflict of interest
The authors have no potential conflicts of interest
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer Statistics, 2008. CA Cancer J Clin 2008; 58:71-96. [PMID 18287387]
- Strimpakos A, Saif MW, Syrigos KN. Pancreatic cancer: from molecular pathogenesis to targeted therapy. Cancer Metastasis Rev 2008; 27:495-522. [PMID 18427734]
- Sugiyama E, Kaniwa N, Kim SR, Kikura-Hanajiri R, Hasegawa R, Maekawa K, et al. Pharmacokinetics of gemcitabine in Japanese cancer patients: the impact of a cytidine deaminase polymorphism. J Clin Oncol 2007; 25:32-42. [PMID 17194903]
- Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy? Br J Cancer 2007; 97:145-51. [PMID 17595663]
- Abraham J, Earl HM, Pharoah PD, Caldas C. Pharmacogenetics of cancer chemotherapy. Biochim Biophys Acta 2006; 1766:168-83. [PMID 17141416]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007; 25:1960-6. [PMID 17452677]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105-11. [PMID 11689955]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res 2007; 67:1030-7. [PMID 17283135]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5:275-84. [PMID 15803154]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003; 100:3983-8. [PMID 12629218]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004; 432:396-401. [PMID 15549107]
- Saif MW. Pancreatic cancer: highlights from the 42nd annual meeting of the American Society of Clinical Oncology, 2006. JOP. J Pancreas (Online) 2006; 7:337-348. [PMID 16832131]


