Keywords
Drug Therapy; everolimus; Neuroendocrine Tumors; Pancreatic Neoplasms; sunitinib
Abbreviations
ATRX: alpha thalassemia/mental retardation syndrome X-linked; CgA: chromogranin A; DAXX: deathdomain- associated protein; NCCN: National Comprehensive Cancer Network; NET: neuroendocrine tumor; pNET: pancreatic neuroendocrine tumors; PS: performance status; RADIANT: RAD001 in Advanced Neuroendocrine Tumors
What Did We Know Before the 2012 ASCO Gastrointestinal Cancers Symposium?
Neuroendocrine tumors (NETs) consist of a diverse group of tumors composed of cells showing neuroendocrine cell differentiation (secretory granules), a subset of which can be further classified by their dominant secretory products. Although it was thought that the neuroendocrine cells that give rise to NETs migrated from the neural crest to the gut endoderm, it is now apparent that enteropancreatic neuroendocrine cells originate from multipotent stem cells that give rise to all epithelial cell types in the gastrointestinal tract and pancreas [1]. NETs show heterogeneity in morphologic, functional and clinical features [2]. Due to the non-uniform nature of this disease reflected by large differences in survival rates with regard to primary tumor site, histologic degree of differentiation and cell type of tumor, it has been challenging to understand the true natural history of NETs. A confounding issue has been the lack of uniform pathological classification or staging system for NETs. At the ASCO GI Cancers Symposium in 2012, several members of the National Comprehensive Cancer Network (NCCN) presented data on the distribution of these tumors at their institutions. In Abstract #187 [3], Choti et al. describe preliminary results from a newly formed database of all adult patients admitted to 7 NCCN institutions with neuroendocrine tumors between the years of 2004 and 2007. They identified 2,542 patients; the majority was diagnosed with carcinoid (51%), the next largest group was diagnosed with pNET (27%). In this sample 53% of patients were women and the median age of diagnosis was 55 years. Use of this database and other national databases will help improve our understanding of the incidence and natural history of this disease.
A subset of NETs involving the pancreas previously termed islet cell tumors or islet cell carcinomas are designated pancreatic NETs (pNETs). Although pNET represent a small percentage of all pancreatic tumors (1.3%), the prevalence of these tumors is significant (9.9% of all pancreatic tumors) and the incidence is increasing [4]. The incidence of pNETs is significantly underestimated in tumor registries including the Surveillance, Epidemiology and End Results (SEER) program which include only malignant neoplasms. Criteria for assessing malignant behavior in pNET include invasion of adjacent organs, regional or distant metastases in addition to tumor mitotic index. In tertiary Oncol ogy centers, the majority of patients with malignant pNETs represent advanced stage tumors with approximately 65% of patients presenting with unresectable or metastatic disease [5]. Prior to 2011, the only approved agent for unresectable disease was streptozocin which was approved prior to 1984 after demonstrating some efficacy in studies in the 1980’s (either alone [6] or in combination with doxorubicin [7]). Further studies have questioned the efficacy of streptozocin [8] and there had not been any new drugs approved in the last 20 years. As a result patients with unresectable pNETs have a poor prognosis. The median survival time for patients with distant metastatic disease is 24 months [5]; the 5-year survival rate of patients with metastatic disease is 30 to 40% [9] and has not changed for 20 years. Several inherited syndromes associated with pNETs including multiple endocrine neoplasia type 1 (MEN1), von Hippel- Lindau disease (vHL), neurofibromatosis 1 (NF1), and the tuberous sclerosis complex are associated with mutations in well-studied oncogenes and tumor suppressor genes that predispose to pNETs, and therefore it is rational to develop therapies targeting these pathways [10]. Unfortunately the underlying genetic abnormalities in these syndromes are relevant in only a subset of the sporadic pNETs [11]. Molecular profiling of pNETs is a critical first step in understanding aberrant regulation of key pathways involved in the initiation and progression of these tumors and defining clinically relevant molecular subgroups that may respond differentially to various targeted treatment protocols. Jiao et al. [12] found the most frequently mutated genes in sporadic pNETs involve proteins involved in chromatin remodeling. In a series of 68 pNETs somatic inactivating mutations in MEN1, which encodes menin, a component of a histone methyltransferase complex involved 44% (30/68), and mutations in genes encoding either of the two subunits of a transcription/chromatin remodeling complex consisting of death-domain-associated protein (DAXX) and alpha thalassemia/mental retardation syndrome X-linked (ATRX) involved 43% (29/68). Mutations in the MEN1 and DAXX/ATRX genes were associated with better prognosis. In addition mutations in genes in the mammalian target of rapamycin (mTOR) pathway were identified in 14% of the tumors. A global gene expression analysis of pNETs revealed that at least two important genes in the mTOR pathway, (TSC2 and PTEN) were downregulated in 85% of primary tumors [13, 14]. Aberrant expression of several tyrosine kinase receptors and overexpression of vascular endothelial growth factor (VEGF) have also been noted in pNET [14]. Utilizing this preclinical data, two targeted agents demonstrated prolongation of progression-free survival in advanced pNET and were approved for this indication in 2011. In the RAD001 in Advanced Neuroendocrine Tumors (RADIANT)-3 trial, an inhibitor of the mTOR pathway, everolimus was superior to placebo in prolonging progression-free survival in patients with unresectable, advanced pNET from 4.6 to 11.0 months [15]. Another phase III trial looked at the multi-kinase inhibitor, sunitinib, in unresectable pNET and found an improvement in progression-free survival from 5.5 to 11.4 months when compared to placebo [16].
What We Learned at the 2012 ASCO Gastrointestinal Cancers Symposium
Post Approval Updates Regarding Therapy with Sunitinib and Everolimus: Refining the Use of Everolimus for Neuroendocrine Tumors
Both sunitinib and everolimus were approved by the Food and Drug Administration (FDA) in 2011 for the treatment of progressive pNET. Both agents were compared to placebo in their respective phase III trials and each showed a significant improvement in the primary outcome of progression-free survival (Table 1). These novel agents have not been evaluated in a direct comparison but post-hoc analysis has been performed comparing the data obtained from these two independent trials. Casciano et al. (Abstract #226 [17]) conducted a cost-effectiveness analysis using a simulated cohort of advanced, progressive pNET patients. The analysis included the cost of the antitumor therapies, other drugs to control symptoms and post-progression therapy in addition to costs associated with physician services, testing, and hospitalization. The model took into account the frequency of adverse events among those who experienced stable disease on either regimen. Using this indirect comparison, the authors estimated that everolimus is associated with an increased cost compared to sunitinib ($12,673 per patient) but that this was associated with a gain in quality adjusted life years (QALYs) of 0.304 years. This results in a cost-effectiveness ratio of $41,702 per QALY which is generally an accepted expense in other Oncol ogy drugs.
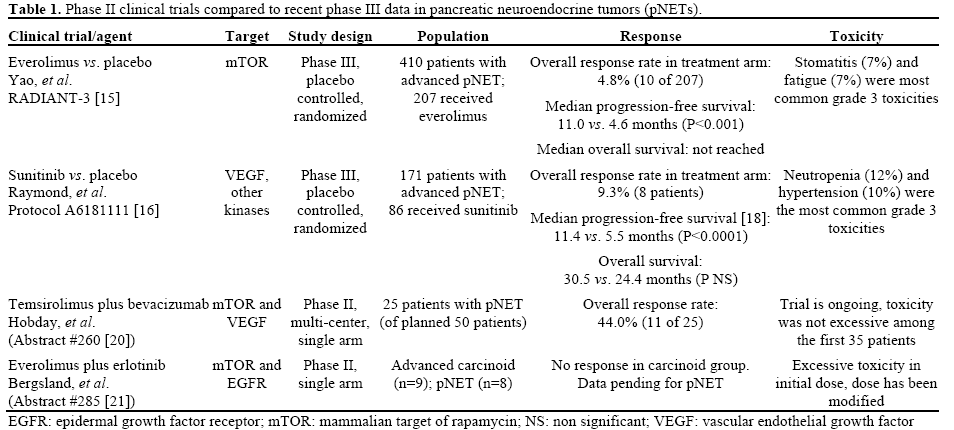
In the phase III trials (summarized in Table 1), each agent demonstrated an improvement in progressionfree survival. In the sunitinib trial [16], data was initially reported on a limited number of deaths (9 in the sunitinib arm and 21 in the placebo arm) and reflected an improvement in overall survival with sunitinib treatment compared to placebo (hazard ratio (HR) for death was 0.41; 95% CI, 0.19 to 0.89; P=0.02); however, the median overall survival was not reached. Updated analysis of events through June 2010 was presented at the ASCO Annual Meeting in 2011 [18] and this revealed a median overall survival of 30.5 months in the sunitinib arm versus 24.4 months in the placebo arm which resulted in a non-significant hazard ration of 0.737 (95% CI, 0.465-1.168; P=0.19). Signorovitch et al. (Abstract #237 [19]) utilized individual patient data from the RADIANT-3 (everolimus) and A6181111 (sunitinib) trials to match patients and compare the outcomes of these trials to each other. The authors matched the patients in the two clinical trials by excluding patients from RADIANT-3 who would not qualify for the A6181111 trial (15 patients with worse performance status) and weighting the patients to match the baseline characteristics of A6181111. After adjusting for this matching, treatment with everolimus was associated with a statistically significant improvement in overall survival compared to the placebo arm of A6181111 (HR for death was 0.61; 95% CI, 0.38-0.98; P=0.04). While there are obvious limitations with this type of post-hoc analysis, the observation is intriguing. No significant difference was seen between the everolimus and sunitinib treated cohorts when these trials were compared.
Novel Therapeutic Combinations for the Management of pNET
With the recent success of the targeted agents, everolimus and sunitinib in prolonging progressionfree survival in pNET, several studies are ongoing to identify potential improvements to this regimen. There were few objective responses in the relevant clinical trials and efforts are underway to find active combinations that result in better tumor response rates. Hobday et al. (Abstract #260 [20]) reported data from an interim analysis of an ongoing phase II trial looking at the combination of an mTOR inhibitor, temsirolimus, and a monoclonal antibody to VEGF, bevacizumab, in progressive pNET. This trial was based on preclinical data that suggests that both of these pathways are upregulated in pNETs and that combined blockade may enhance tumor response. All patients had well or moderately differentiated progressive pNET, performance status (PS) of 0 or 1, and no prior exposure to either targeted agent. This interim analysis was planned after enrollment of 25 of the total 50 patients. Among the first 25 patients, partial response was documented in 44% of patients (11 of 25) which compares favorably to the 10% response rate seen in the phase III trial of everolimus alone and warrants continuation of this study. Toxicity data was also reported from the first 35 enrolled patients and demonstrated that the regimen was well tolerated; this study is continuing to accrue patients.
Another phase II trial was reported by Bergsland et al. (Abstract #285 [21]) and looked at the combination of the mTOR inhibitor everolimus, with a small molecule inhibitor of the epidermal growth factor receptor (EGFR), erlotinib. Based on preclinical data this combination is also thought to lead to enhanced tumor response compared to either individual agent. The study included patients with well to moderately differentiated NETs and it was divided between patients with low-grade carcinoids (n=9) and pNET (n=8). The initial dose utilized was everolimus 5 mg daily and erlotinib 150 mg daily; however 3 of the first 7 patients experienced grade 3 toxicity (stomatitis) requiring a dose reduction of erlotinib to 100 mg daily. Ten additional patients have been treated at this dose level with only one grade 3 toxicity (10%) to date. Among the 9 patients with carcinoid there were no objective responses and enrollment to this cohort has been stopped. The efficacy analysis of the pNET cohort has not yet been reported.
Identifying Biomarkers in pNET
The heterogeneity of neuroendocrine tumors is well described. There is a need to identify prognostic biomarkers and to identify characteristics that may predict response to therapy. Yao et al. (Abstract #157 [22]) presented data from a multivariate analysis of the large randomized, phase III, RADIANT-2 trial. In this trial, 429 patients with neuroendocrine tumors of diverse sites including a small number of pNETs (6%) were treated with long acting octreotide in addition to either everolimus or placebo. Treatment with everolimus was associated with a prolongation in progression-free survival compared to placebo. In this abstract the authors reviewed many variables in this study and identified several prognostic factors. Nonelevated levels of chromogranin A (CgA) or the serotonin metabolite 5-hydroxyindoleacetic acid (5- HIAA) were associated with increased time to progression. Other important factors were the presence of liver metastasis or a primary site in the lung, which were associated with shortened time to progression. On multivariate analysis, non-elevated level of CgA at baseline was associated with reduced rate of progression (HR: 0.47; P<0.001), as was patient functional status with patients who are ambulatory and active (World Health Organization (WHO) PS (performance status) of 1 or 0) demonstrating a reduced rate of progression compared to those with more limitations (WHO PS of 2) (HR: 0.69; P=0.006). Bone involvement and lung as a primary site were associated with increased rates of progression (HR: 1.52; P=0.02 and HR: 1.55, respectively; P=0.04).
A retrospective study reported by Alistar et al. (Abstract #166 [23]) looked at a series of 142 patients who underwent surgical resection of a pNET between 1980 and 2011. The authors looked at serum levels of CgA and serotonin in the post-operative period in these patients to determine if these markers predict survival. There was no correlation between normal serum levels and survival among the 27 patients who had CgA measured in the immediate post-operative period (0-3 months). However, in the 30 patients who had CgA measured in the late post-operative period (3-12 months), there was a non-significant trend to improvement in survival among those who had a normal CgA level (P=0.14 for survival). Serotonin levels were not found to correlate with survival in this patient population when measured in the post-operative period.
Discussion
NETs are increasing in incidence. Perhaps in part the increased incidence is due to improvements in diagnostic imaging with detection of smaller lesions in addition to the incidental diagnosis of asymptomatic cases [24]. For the group of malignant NET, effective treatment options will need to be identified through well designed clinical trials. A collaboration of NCCN sites is underway [3] to improve our characterization of these patients. It has been observed that pNETs represent a significant proportion of NETs and account for a growing proportion of pancreatic tumors. Recently two targeted agents have been approved by the FDA for the treatment of progressive pNET. Everolimus is an mTOR inhibitor, which inhibits cell growth, proliferation, and angiogenesis. Sunitinib is a multi-kinase inhibitor that is thought to have an effect in pNET through inhibition of VEGF, which plays a role in angiogenesis in pNET. As summarized above, several abstracts presented at the 2012 ASCO Gastrointestinal Cancers Symposium contribute important information to this field. Though direct clinical studies do not address the role of sunitinib versus everolimus in advanced pNET, the costeffectiveness analysis performed by Casciano et al. (Abstract #226 [17]) suggests that everolimus may provide an incremental gain in quality adjusted life years at a reasonable cost ($41,702/QALY). Similar analysis of the two trials by Signorovitch et al. (Abstract #237 [19]) suggests that there is no significant difference in overall or progression-free survival between the everolimus and sunitinib cohorts but that everolimus is associated with improved survival when compared to the placebo arm of the sunitinib trial. As stated above, such a post-hoc analysis must be interpreted with caution. Combination therapy utilizing agents targeting multiple pathways are ongoing, and interim data from Hobday et al. (Abstract #260 [20]) finds support for the combination of an mTOR inhibitor (temsirolimus) with blockade of the VEGF pathway (bevacizumab). The combination of everolimus and erlotinib appears to have excessive toxicity although final analysis in pNET has not been reported [21]. Analysis of the RADIANT-2 trial suggests that a non-elevated baseline chromogranin level is a favorable prognostic indicator, while the presence of bone involvement or lung as a primary site is a poor prognostic factor. Finally, an abstract by Yamaguchi et al. (Abstract #274 [25]) addressed the role of therapy for the more complicated cases of poorly differentiated neuroendocrine tumors. These tumors are more aggressive than carcinoids and pNETs and the optimal chemotherapeutic regimen is not known. Phase II data suggests that combination therapy with cisplatin plus either etoposide [26] or irinotecan [27] is associated with responses. Yamaguchi et al. (Abstract #274 [25]) reported on a large series of patients treated for poorly differentiated NET in 23 centers in Japan. They identified 258 patients who had high risk tumors; namely poorly differentiated NET (PDNET), small cell carcinoma, or mixed exocrineendocrine tumors with a PDNET component. All patients selected had inoperable or recurrent disease with a primary tumor in the gastrointestinal tract or pancreas. The median age at diagnosis was 62 years and the most common treatment was combination irinotecan and cisplatin. The response rate to this regimen was substantial; 51% in the tumors of GI origin, 39% in those of hepatobiliary/pancreatic origin. Median overall survival in these two groups was 13.4 and 10.1 months, respectively. Responses were also seen with the combination of etoposide and cisplatin, though with this regimen as well overall survival was longer in tumors of GI origin as compared to hepatobiliary/ pancreatic origin.
In summary, the abstracts presented at the 2012 ASCO Gastrointestinal Cancers Symposium highlight the continued advances in understanding the epidemiology, prognostic factors, and novel treatments to improve care of advanced pancreatic neuroendocrine tumors. With a number of new active agents identified, and new trials testing logical combinations, the future looks promising for patients suffering from this rare form of pancreatic and GI cancer.
Conflicts of interest
The authors have no conflicts to disclose
References
- Barker N, van Es JH, Kuipers J, et al. Identification of stem cellsin small intestine and colon by marker gene Lgr5. Nature2007;449:1003-7.
- Jensen RT. Carcinoid tumors and the carcinoid syndrome. In: DeVita VTJ, Lawrence, T., Rosenberg, S.A., ed. Cancer: Principles and Practice of Oncol ogy. Philadelphia: Lippincott, Williams, and Wilkins; 2008.
- Choti MA, Mayorga MA, Bobiak S, et al. Baseline demographics of patients with neuroendocrine tumors presenting toseven National Comprehensive Cancer Network (NCCN) institutions: Development of a multi-institutional outcomes database.J Clin Oncol 30, 2012 (suppl 4;abstr 187).
- Yao JC, Eisner MP, Leary C, et al. Population-based study ofislet cell carcinoma. Ann Surg Oncol 2007;14:3492-500.
- Yao JC, Hassan M, Phan A, et al. One hundred years after"carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008;26:3063-72.
- Broder LE, Carter SK. Pancreatic islet cell carcinoma. II. Resultsof therapy with streptozotocin in 52 patients. Ann Intern Med1973;79:108-18.
- Moertel CG, Lefkopoulo M, Lipsitz S, Hahn RG, Klaassen D.Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocinin the treatment of advanced islet-cell carcinoma. N Engl J Med1992;326:519-23.
- McCollum AD, Kulke MH, Ryan DP, et al. Lack of efficacy ofstreptozocin and doxorubicin in patients with advanced pancreatic endocrine tumors. Am J Clin Oncol 2004;27:485-8.
- Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology 2008;135:1469-92.
- Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer2008;113:1807-43.
- Zikusoka MN, Kidd M, Eick G, Latich I, Modlin IM. Themolecular genetics of gastro entero pancreatic neuroendocrine tumors.Cancer 2005;104:2292-309.
- Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuro endocrine tumors. Science 2011;331:1199-203.
- Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrinetumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol 2010;28:245-55.
- Zhang J, Jia Z, Li Q, et al. Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis anddecreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer 2007;109:1478-86.
- Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuro endocrine tumors. N Engl J Med 2011;364:514-23.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for thetreatment of pancreatic neuroendocrine tumors. N Engl J Med2011;364:501-13.
- Casciano R, Chulikavit M, Perrin A, Liu Z, Wang X, Babigumira J, Garrison LP. Cost-effectiveness of treating patientswith advanced progressive pancreatic neuroendocrine tumor witheverolimus versus sunitinib in the United States. J Clin Oncol 2012;30(Suppl 4):Abstract 226.
- Raymond E, NP, Raoul J, et al. Updated overall survival (OS)and progression-free survival (PFS) by blinded independent centralreview (BICR) of sunitinib (SU) versus placebo (PBO) for patientwith advanced unresectable neuroendocrine tumors (NET). J Clin Oncol 2011;29(Suppl.):4008.
- Signorovitch J, Swallow E, Kantor E, et al. Overall survival with everolimus, sunitinb, and placebo for advanced pancreatic neuroendocrine tumors: A matching-adjusted indirect comparison of randomized trials. J Clin Oncol ;30, 2012 (suppl 4; abstr 237).
- Hobday TJ, Qin R, Reidy DL, Moore MJ, Strosberg JR, Kaubisch A, et al. Multicenter phase II trial of temsirolimus (TEM) and bevacizumab (BEV) in pancreatic neuroendocrine tumor (PNET). J Clin Oncol 2012; 30(Suppl 4):Abstract 260.
- Bergsland EK, Watt L, Ko AH, Tempero MA, Korn WK, KelleyRK, et al. A phase II study to evaluate the safety and efficacy ofRAD001 plus erlotinib in patients with well-differentiated neuro endocrine tumors (NET). J Clin Oncol 2012; 30(Suppl4):Abstract 285.
- Yao JC, Hainsworth J, Wolin EM, et al. Multivariate analysis including biomarkers in the phase III RADIANT-2 study of octreotide LAR plus everolimus (E+O) or placebo (P+O) amongpatients with advanced neuroendocrine tumors (NET). J Clin Oncol 30, 2012 (suppl 4;abstr 157).
- Alistar AT, Warner R, Moshier E, et al. Biomarkers inpancreatic neuroendocrine tumors. J Clin Oncol 30, 2012 (suppl4;abstr 166).
- Bruzoni M, Johnston E, Sasson AR. Pancreatic incidentalomas:clinical and pathologic spectrum. Am J Surg 2008;195:329-32;discussion 32.
- Yamaguchi T, Machida N, Kasuga A, Takahashi H, Sudo K,Nishina T, et al. Multicenter retrospective analysis of systemic chemotherapy in poorly differentiated neuroendocrine carcinoma ofthe digestive system. J Clin Oncol 2012; 30(Suppl 4):Abstract 274.
- Mitry E, Baudin E, Ducreux M, et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin.Br J Cancer 1999;81:1351-5.27.
- Kulke MH, Wu B, Ryan DP, et al. A phase II trial of irinotecanand cisplatin in patients with metastatic neuroendocrine tumors. DigDis Sci 2006;51:1033-8.

