Keywords
Endoscopy; Laparoscopy; Pancreatic Pseudocyst
Abbrevations
SPT solid pseudopapillary tumors
INTRODUCTION
A review of the National Cancer Institute SEER (Surveillance, Epidemiology, and End Results) Publicuse Database from 1973 to 2004 found an incidence of malignant pancreatic cancer in patients less than 30 years to be 0.46/million [1]. The numbers for pediatric pancreatic cancer and tumors are much smaller accounting for less than 0.2% of pediatric cancer-related deaths [2]. Needless to say, standard of care for pediatric pancreatic tumors has yet to be determined.
METHODS
We performed an extensive review of the English Language literature focusing on pediatric pancreatic malignancies. We primarily used the PubMed search engine to access the Medline database but performed secondary searches using EMBASE, MEDLINE and SCOPUS. All articles in English, including case reports, involving patients under the age of 18 years were included.
However we have not quoted extensively from such case reports focusing the review predominantly on data from larger case series. While this was sufficient in a majority of cases, in tumors such as Intraductal Papillary Mucinous neoplasms (IPMN) only two case reports were available for the pediatric population. While we have alluded to this for the sake of completeness, the management of these rare tumors should be multi disciplinary. The articles reviewed (Table-1) were not time limited as pathologic classification for these tumors have evolved, however priority was given to articles published after 1995.
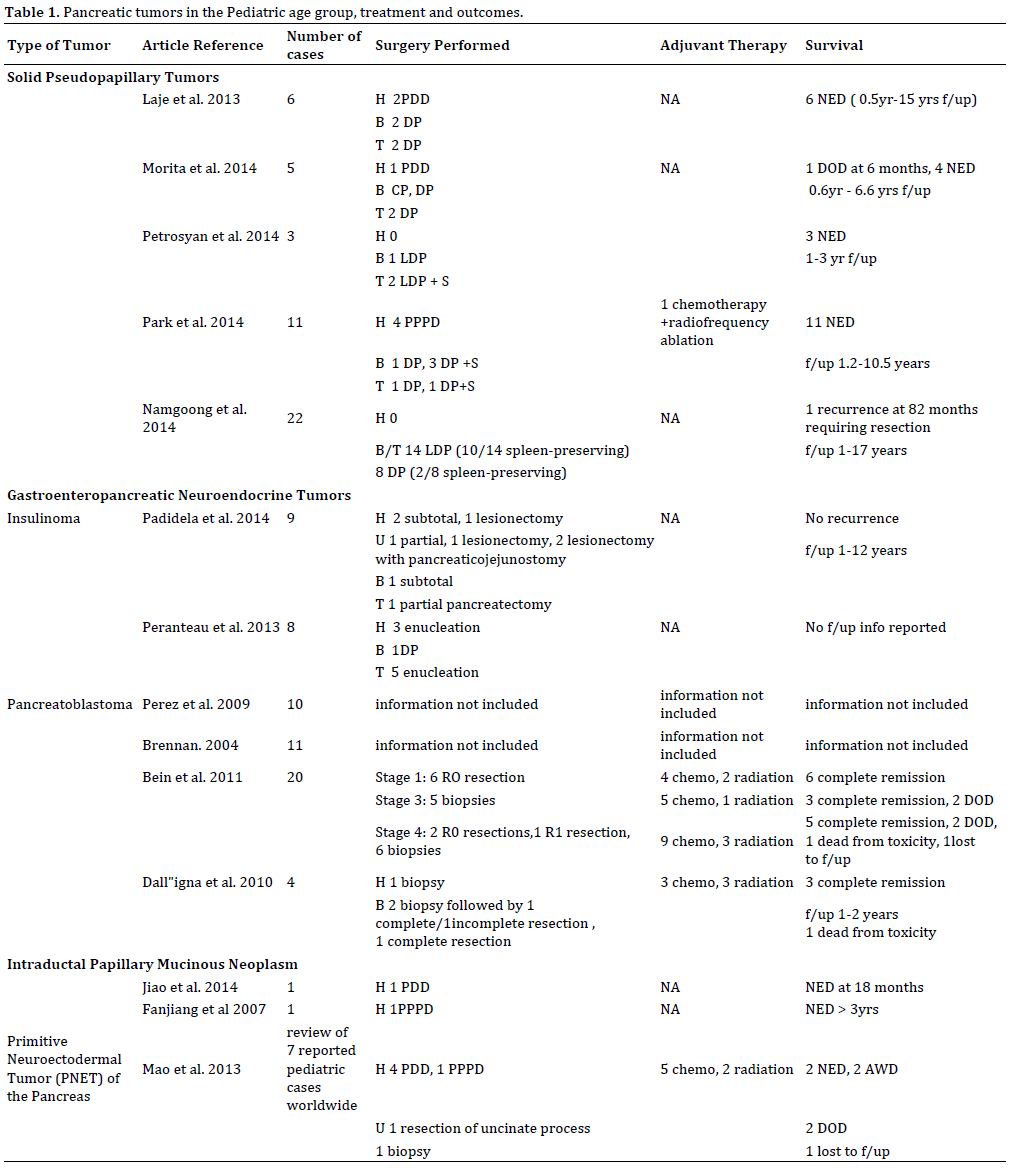
In this review we have held to the original basis of classification of pancreatic tumors based on cell of origin as Exocrine or Endocrine. Exocrine tumors were further classified as epithelial or non-epithelial. Epithelial pancreatic tumors, such as solid pseudopapillary tumor (SPT), pancreatoblastoma, acinar carcinoma, and ductal carcinoma arise from ductal and acinar cells or cells of unknown origin are more common than nonepithelial tumors such as lymphomas, primitive neuroectodermal tumors, and mesenchymal tumors [2]. Endocrine tumors are also known as Gastroenteropancreatic neuroendocrine tumors (GEP-NET) and are further subclassified as insulinoma, gastrinoma, glucagonoma, and VIPOMA [2].
We have elaborated on the pathology and evaluation of these tumors and have tried to specifically address the controversies associated with management of these tumors including 1). Type of surgery: open or laparoscopic? 2). Surgery as a diagnostic procedure 3). Should a biopsy precede the complete excision? 4). If surgery is a distal pancreatectomy, should the spleen be preserved? 5). Are the chances of splenic conservation higher with an open versus laparoscopic procedure?
RESULTS
Solid Pseudopapillary Tumors
Solid Pseudopapillary Tumors (SPT) were first described in 1959 by Frantz and subsequently classified in 1996 by the WHO [3, 4]. These tumors were previously known as papillary cystic epithelial neoplasms, solid/ papillary neoplasms, papillary cystic tumors, and solid/ cystic tumors [5]. Typically, SPT are found in young females in the second or third decade of life [5]. Female to male ratio has been reported ranging from 2:1 to 10:1 [6]. Pathology reveals ovoid nuclei and eosinophilic granules arranged in sheets with pseudopapillary architecture [5]. In one case series, all tumors were positive for a particular dot-like staining pattern of CD-99. [8] They represent 2-3% of all pancreatic tumors [5]. There are no tumor markers specific to SPT. One series showed elevated alpha fetal protein (AFP), carcinoembryonic antigen (CEA), cancer antigen 19-9 (CA 19-9), and cancer antigen 125 (CA 125) in some patients with SPT but not all [5].
Diagnosis
SPT in children are typically found as incidental masses given the benign, indolent nature of these tumors [5]. Typical presenting symptom is abdominal pain [5, 7]. Though rare, there have been reported cases of hemoperitoneum secondary to traumatic or spontaneous rupture of SPT in children. Multiple imaging modalities have been used to identify these lesions, including abdominal ultrasound (US), computed tomography (CT), and magnetic resonance imaging (MRI). Ozcan and colleagues reported on 15 patients diagnosed with primary pancreatic tumors with imaging over a 22-year period from 1991 to 2013. 7 of the 15 patients had SPT seen on US, CT, or MRI. Tumor size ranged from 2 to 10 cm. On US, most tumors showed heterogeneity with a mixture of solid and cystic components with some vascularity. 6 of the 7 showed encapsulation. All tumors on CT had contrast enhancement. MRI for one of the tumors showed areas of possible hemorrhage inside the tumor. No metastases were seen on any modality of imaging.
Treatment
Biopsy of these tumors is generally not recommended given the risk of recurrence even after complete resection [3]. There is general consensus that surgical resection offers the only cure. The type of pancreatic resection is dependent on location of the tumor. Because these patients are children there is the dual purpose of complete excision of the tumor with functional preservation of the pancreas if at all possible. Enucleation for small SPT has been done in the past. Pathologic studies of SPTs reported in a case series by Morita et al. [4] show that despite well-circumscribed tumors seen on imaging, there was incomplete tumor capsule in 4 out of 5 SPTs, with local infiltration into surrounding parenchyma and pancreatic duct. Simple enucleation has a reported increased risk of local recurrence probably due to incomplete resection [7]. Ductal involvement with incomplete resection may also lead to the post-operative morbidity of a pancreatic fistula.
Even though, the first laparoscopic distal pancreatectomy in a porcine model was described in 1994 and the first adult case report was published two years later [8], it was not until 2003 that the first successful LDP in a 9-year old boy was reported from France [9]. The reasons for the delay in adopting laparoscopic technique for distal pancreatectomy in children are two-fold. First, given the rarity of the disease process, it is difficult for pediatric surgeons to acquire the numbers for the learning curve. Second, the delay in developing instruments and technology suitable for the children smaller physical size has led to a delay in adopting laparoscopic technique in the pediatric population compared to the adult population [8]. However, as tool are being developed, laparoscopic distal pancreatectomies for benign lesions in children are not so rare. Namgoong et al. [8] reports the largest case series of 22 patients with SPT who underwent distal pancreatectomies, 14 laparoscopic (LDP) and 8 open (ODP). The data was collected from 1995 to 2012. From 2007 on, distal pancreatectomies were performed laparoscopically. The results of the case series showed that patients in the LDP group had shorter recovery time in terms of length of hospitalization and time to oral intake, shorter operative time, and less blood loss compared to the ODP group. However, there were larger tumors with a high incidence of metastases in the ODP group that could have adversely affected all the aforementioned outcomes. This study reported a higher incidence of spleen preserving distal pancreatectomies in the LDP group. Postoperative morbidities were similar in each group with pancreatic fistulas being the more common one. None of the complications required re-operation. There was only one recurrence on the LDP at 82 months requiring reoperation and resection and reconstruction of the portal vein. This study did not show superiority of LDP over ODP, but it did show that LDP is safe and feasible for children with SPT in the tail of the pancreas.
Survival
SPT generally considered benign lesions. However, they do have low grade malignant potential with metastases found 19.5% of the time when discovered [5]. Adults with SPT have a higher percentage of malignancy than children diagnosed with SPT [9]. Greater than 95% of SPT are localized to the pancreas. Recurrence rate after resection has been reported to be approximately 10%. [5] The 5-year survival rate has been reported to be 95- 98% after complete resection.[5] Prognosis for this type of tumor is generally considered very good with appropriate treatment.
Gastroenteropancreatic Neuroendocrine Tumors (GEP-NET)
GEP-NETs are slow growing tumors that represent 1-2% of all pancreatic tumors [10]. They were first described as carcinoid tumors in 1907 and can be associated with familial syndromes such as multiple endocrine neoplasia (MEN) 1/2, Von Hippel-Lindau (VHL), and tuberous sclerosis [10]. One-third of GEP-NETS are found in the pancreas. Functioning tumors can produce symptoms specific to the substance secreted such as hypoglycemia in insulinomas. Insulinomas are the most common type found in children. While there were two cases of VIPOMAs reported in children, in this review we will focus on insulinomas, the most common type of pediatric tumor.
Insulinoma
Insulinomas have an estimated incidence of 1 in 4 million people. They are more often benign lesions with a 6% incidence of malignancy [11]. Ten percent are associated with multiple endocrine neoplasia-1 (MEN-1) [11].
Diagnosis
Symptoms of Insulinoma are typical of hypoglycemia: hunger, sweating, anxiety, behavioral changes, seizures, palpitations. If Whipple’s triad (signs of hypoglycemia, low measured plasma glucose, and resolution of symptoms with glucose consumption) is present, insulinoma should be suspected. An important differential diagnosis is Congenital Hyperinsulinism (CHI) that is a cause of hyperinsulinemic hypoglycemia in children that occurs more commonly in infancy. Nesidioblastosis is the cause of CHI [11]. The annual incidence 1 in 50,000 births in sporadic forms and as high as 1 in 2500 births in familial disease [11]. Histopathologic findings are hypertrophic beta cells and islets [11]. There are diffuse and focal forms which determine whether treatment is partial or near-complete pancreatectomy [11]. If hypoglycemia symptoms arise later in childhood, insulinoma should be suspected. If diagnosis between CHI and Insulinoma is difficult, maximum hepatic venous insulin (mHVI) and relative hepatic venous insulin (rHVI) measurements after selective arterial calcium injection may help in distinguishing between the two [12]. Insulinoma in childhood is a rare entity with the two largest case series consisting of eight and seven patients [11, 13]. There is often a delay in diagnosis from onset of symptoms most likely because children are unable to dependably recognize and communicate their symptoms. In one series, younger children, 10 or younger, had a longer duration of symptoms before diagnosis compared to the older children with insulinoma [13]. Some children who were later diagnosed with insulinoma are initially diagnosed and treated for seizure disorders [13].
The most common imaging techniques utilized for localizing the insulinoma include CT, abdominal ultrasound, MRI, and PET-CT. The most successful noninvasive imaging was MRI identifying 7/8 lesions in one series and 3/6 in another 9 [11, 13]. PET-CT was able to identify the missed lesion on MRI in one study [11]. In the other series by Peranteau et al. PET-CT identified 3/3 lesions when used, while more invasive studies such as arterial stimulus with venous sampling (ASVS) and transhepatic portal venous sampling (THVS) identified the presence of insulinoma when non-invasive imaging had failed [13].
Treatment
All patients in the two large series that were reviewed underwent exploratory laparotomy [11, 13]. In the series by Peranteau et al. all except one lesion was identified by palpation [13]. Endoscopic ultrasound was able to identify the non-palpable lesion [13]. Surgery is the mainstay of treatment for insulinoma. Size of the tumors in both studies ranged from 0.7 cm to 2cm [11, 13]. Insulinomas were not found to be predominantly in one section of the pancreas versus another [11, 13]. When possible enucleation was preferred given the benign nature of these lesions [13]. However, in the case series by Padidela et al. of patients with proximal lesions in the head or the uncinate process (6/9), only one underwent enucleation, while the others underwent subtotal pancreatectomy or lesionectomy with pancreatico-jejunostomy [11]. Three patients in these two series combined were positive for MEN-1 [11, 13].
Two pancreatic fistulas and one take back for missed insulinoma in the splenic hilum were reported as postoperative complications [13]. Both fistulas required ERCP and pancreatic stent placement. One failed that management and required a distal pancreatectomy and over sewing of the resection line [13]. The other series out of the United Kingdom did not report post-operative complication [11].
Survival
The majority of insulinomas are benign with only a 6% incidence of malignancy, and long term survival for nonmalignant disease following surgical resection is around 80-90% in adults and is estimated to be the same in children. Post-operative surveillance is important, especially for the patients with MEN-1. In adults, the risk of recurrence is higher in patients with MEN-1 at ten years than without (21% vs. 5%) [11, 13].
Summary
For localization of insulinoma, MRI should be used as first-line imaging with or without endoscopic ultrasound. If lesions are not identified on MRI, PET-CT scans may successfully identify the location of the Insulinoma. Given the benign nature of these lesions, pancreas-sparing surgery is the preferred operative technique. Given the small numbers of cases in the literature reviewed, it was difficult to assess the development of post-operative pancreatic fistulas with use of enucleation technique over partial/subtotal pancreatectomy. Protocols to decrease post-operative pancreatic fistula formation include use of tissue sealants and mattress sutures to reinforce the site of enucleation [13]. If there is a concern for ductal involvement at the time of surgery, partial pancreatectomy should be performed [13].
Pancreatoblastoma (PBL)
PBL is the most common malignant pancreatic tumor in children. PBL was first coined in 1977. Prior to that it was known as infantile carcinoma of the pancreas or juvenile adenocarcinoma of the pancreas [14]. It typically affects younger children, ages 1-8 with a mean age of 5 [14]. These tumors are thought to arise from persistent embryonic pancreatic acinar cells [14].
Diagnosis
Common presenting symptoms of PBL are abdominal pain and palpable epigastric mass, while jaundice and emesis from obstruction is less common [14-18]. Neonatal cases are often associated with Beckwith-Wieddemann Syndrome or Familial Adenomatous Polyposis [14]. Cross sectional imaging is important to exclude porta hepatis and vascular invasion that would contraindicate surgical intervention.
Surgery
While the majority of PBL has been reported as single case reports, there have been a few case series published over the last 15 years to give an idea of the rarity of the disease and an overview of treatment and prognosis [14-18]. The largest case series has come out of the European Cooperative Study Group for Pediatric Rare Tumors (EXPeRT) with 20 cases of PBL over a nine-year review period from 2000-2009 [16]. There were 10 and 11 PBL cases from the North American population based Surveillance, Epidemiology and End Results (SEER) database from 1973 to 2004 and the United Kingdom National Registry of Childhood Tumours, respectively [17, 18].
Surgical resection is the mainstay of treatment for PBL whether at initial diagnosis or after chemotherapy [16]. If at initial laparotomy, complete resection is deemed unfeasible, an open biopsy must be performed for diagnosis. Characteristics of unresectable disease include invasion of one of two structures of the porta hepatis: the portal vein or hepatic artery, metastatic disease, local invasion into nearby major vasculature: the aorta, inferior vena cava, or celiac artery [16, 18].
Survival
In the EXPeRT case series all cases of complete resection at time of initial laparotomy without previous chemotherapy survived at the end of the review period [16]. Metastatic disease that had response to chemotherapy allowing for subsequent complete resection, even some cases with metastatectomy included, had good prognosis [16]. For the whole series, 5-year event-free survival and overall survival were 58.8% and 79.4%, respectively. Outcome did not correlate with tumor site and size, but was strongly influenced by the feasibility of tumor complete resection. Survival was not related to size of the tumor, but to complete resection [16, 18].
Intraductal Papillary Mucinous Neoplasm (IPMN)
IPMN is a premalignant lesion of the pancreas [19]. It typically occurs in males in the sixth decade of life and at the head of pancreas and has been reported with greater frequency after the 1990’s. Presenting symptoms are usually a history of recurrent pancreatitis. At time of discovery, 50% of the patients present with invasive carcinoma.
Diagnosis and Surgery
There have been only two case reports of IPMN in pediatric patients. The first case report was of a 14-year old male who originally presented with epigastric pain for one year. Initial computed tomography (CT) of the abdomen showed an enlarged pancreatic head. Evaluation one year later for persistent symptoms with ERCP showed a characteristic fish-eye appearance of the ampulla for IPMN. Repeat CT showed a 13 mm hypodense lesion in the head of the pancreas. The patient underwent a pyloruspreserving pancreatoduodenectomy. He has done well for over three years without evidence of recurrence [20].
The youngest reported patient with IPMN was a newborn full-term female infant reported by Jiao and colleagues in 2015 in Pancreatology [21]. Presenting symptoms were hypoglycemia one day after discharge from the hospital after birth. Ultrasound showed a cystic mass next to the left kidney. MRI revealed a 2.2x3.7x3.9 cm cystic mass in the head of the pancreas. The neonate underwent a Whipple procedure for surgical resection. Pathology confirmed a 4.0cm IPMN with high grade dysplasia and enlarged islets of Langerhans. KRAS mutation was found only in the tumor cells, and a de novo germline SKIL mutation was also found upon further investigation. This was the first case of IPMN and concurrent congenital hyperinsulinism in an infant.
Survival
Survival reports can only be extrapolated from adult case series as no substantial pediatric data is available. 3-year survival after surgical resection is reported to be 60-80%. It is decreased to 21% with invasive cancer.
Primitive Neuroectodermal Tumor (PNET) of the Pancreas
PNETs are small round cell tumors that were first described in 1918 in association with peripheral nerves. They are part of the Ewing sarcoma family of tumors and typically found in soft tissue of the pelvis or extremities.
Diagnosis and Surgery
Mao et al. reported the first case of pancreatic PNET in Asia and the 14th case reported in the literature [22]. Age range for all 14 patients was from 6-31 years of age, with 7 of the 14 under the age of 18 years [22]. These patients presented more commonly with jaundice from biliary obstruction or abdominal pain from mass effect. Tumors from the pediatric patients were typically CD99/O13 + and positive for the t 11; 22(q24;q12) chromosomal translocation [23]. Treatment should be aggressive with complete surgical resection if possible. Chemotherapy and radiation therapy may extend survival than just surgical resection alone [22].
Survival
PNET tumors are highly aggressive with very poor prognosis [22, 25, 26]. Five year survival for PNET is reported to be 50%. In the series reported above, longest follow-up was 50 months and probably the longest survival as well [22].
Pancreatic Ductal and Acinar Carcinoma
Acinar cell carcinoma typically affects men in their fifth decade of life, while ductal carcinoma is rare in patients under the age of 40 [1]. Pediatric cases have been reported for both types of cancers but are extremely rare [1, 26]. Most reported cases of ductal carcinoma in the pediatric population have been before pathologic diagnosis of SPT or pancreatoblastoma, so many cases were likely misidentified as ductal carcinoma [26]. Surgery is still the mainstay of treatment, regardless of type of tumor. Evaluation and surgical treatment needs to be individualized and no recommendations can be given based on the sparse pediatric literature on this topic. Complete resection of acinar carcinoma has led to survival without recurrence [1].
CONCLUSIONS
SPT and insulinomas are the common pediatric pancreatic tumors. As they are often benign, a laparoscopic approach to resection has been shown to be acceptable in some case series. Malignant pancreatic tumors in the pediatric population can be very aggressive. They are treated with complete resection, sometimes in association with chemotherapy and radiation therapy, with the intent of extending disease-free survival. Individual countries have formed databases for these rare tumors with the purpose of elucidating presentation, diagnosis, and treatment of these rare tumors. An international collaborative would be ideal to capture potential missed cases.
Conflict of Interest
The authors declare that they have no competing interests.
References
- Brecht IB, Schneider DT, Klöppel G, von Schweinitz D, Barthlen W, Hamre MR. Malignant pancreatic tumors in children and young adults: evaluation of 228 patients identified through the Surveillance, Epidemiology, and End Result (SEER) database. Klin Padiatr 2011; 223:341-5. [PMID: 22012608]
- Yu DC, Kozakewich HP, Perez-Atayde AR, Shamberger RC, Weldon CB. Childhood pancreatic tumors: a single institution experience. J Pediatr Surg 2009; 44:2267-72. [PMID: 20006007]
- Park JY, Kim SG, Park J. Solid pseudopapillary tumor of the pancreas in children: 15-year experience at a single institution with assays using an immunohistochemical panel. Ann Surg Treat Res 2014; 86:130-5. [PMID: 24761421]
- Shet NS, Cole BL, Iyer RS. Imaging of pediatric pancreatic neoplasms with radiologic-histopathologic correlation. AJR Am J Roentgenol 2014; 202:1337-48. [PMID: 24848833]
- Morita K, Urushihara N, Fukumoto K, Miyano G, Yamoto M, Nouso H, Miyake H, Kaneshiro M. Solid pseudopapillary tumor of the pancreas in children: surgical intervention strategies based on pathological findings. Pediatr Surg Int 2014; 30:253-7. [PMID: 24442211]
- Laje P, Bhatti TR, Adzick NS. Solid pseudopapillary neoplasm of the pancreas in children: a 15-year experience and the identification of a unique immunohistochemical marker. J Pediatr Surg 2013; 48:2054-60. [PMID: 24094957]
- Namgoong JM, Kim DY, Kim SC, Kim SC, Hwang JH, Song KB. Laparoscopic distal pancreatectomy to treat solid pseudopapillary tumors in children: transition from open to laparoscopic approaches in suitable cases. Pediatr Surg Int. 2014; 30:259-66. [PMID: 24468715]
- Service FJ, McMahon MM, O'Brien PC, Ballard DJ. Functioning insulinoma--incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 1991; 66:711-9. [PMID: 1677058]
- Kaczirek K, Niederle B. Nesidioblastosis: an old term and a new understanding. World J Surg 2004; 28:1227-30. [PMID: 15517495]
- Shorter NA, Glick RD, Klimstra DS, Brennan MF, Laquaglia MP. Malignant pancreatic tumors in childhood and adolescence: The Memorial Sloan-Kettering experience, 1967 to present. J Pediatr Surg 2002; 37:887-92. [PMID: 12037756]
- Padidela R, Fiest M, Arya V, Smith VV, Ashworth M, Rampling D, Newbould M, Batra G, et al. Insulinoma in childhood: clinical, radiological, molecular and histological aspects of nine patients. Eur J Endocrinol 2014; 170:741-7. [PMID: 24599222]
- Thompson SM, Vella A, Thompson GB, Rumilla KM, Service FJ, Grant CS, Andrews JC. Selective arterial calcium stimulation with hepatic venous sampling differentiates insulinoma from nesidioblastosis. J Clin Endocrinol Metab 2015;100:4189-97.
- Peranteau WH, Palladino AA, Bhatti TR, Becker SA, States LJ, Stanley CA, Adzick NS. The surgical management of insulinomas in children. J Pediatr Surg 2013; 48:2517-24. [PMID: 24314196]
- Glick RD, Pashankar FD, Pappo A, Laquaglia MP. Management of pancreatoblastoma in children and young adults. J Pediatr Hematol Oncol 2012; 2:S47-50. [PMID: 22525406]
- Dall'igna P, Cecchetto G, Bisogno G, Conte M, Chiesa PL, D'Angelo P, De Leonardis F, et al. Pancreatic tumors in children and adolescents: the Italian TREP project experience. Pediatr Blood Cancer 2010; 54:675-80. [PMID: 19998473]
- Bien E, Godzinski J, Dall'igna P, Defachelles AS, Stachowicz-Stencel T, Orbach D, Bisogno G, et al. Pancreatoblastoma: a report from the European cooperative study group for paediatric rare tumours (EXPeRT). Eur J Cancer 2011; 47:2347-52. [PMID: 21696948]
- Brennan B. Pancreatoblastoma. August 2004. www.orpha.net/data/patho/GB/uk-pancrea.pdf.
- Perez EA, Gutierrez JC, Koniaris LG, Neville HL, Thompson WR, Sola JE. Malignant pancreatic tumors: incidence and outcome in 58 pediatric patients. J Pediatr Surg 2009; 44:197-203. [PMID: 191597]
- Ohashi K, Murakami Y, Murayama M, Takekoshi T, Ohta H, Ohashi I, et al. Four cases of mucus secreting pancreatic cancer. Prog Dig Endoscopy 1982; 20:348-51
- Fanjiang G, Guelrud M, Gupta M, Dayal Y, Katz AJ. Intraductal papillary-mucinous neoplasm of the pancreas in a 14-year-old. J Pediatr Gastroenterol Nutr 2007; 44:287-90. [PMID: 17255848]
- Jiao Y, Lumpkins K, Terhune J, Hruban RH, Klein A, Kinzler KW, Papadopoulos N, et al. Intraductal papillary mucinous neoplasm in a neonate with congenital hyperinsulinism and a de novo germline SKIL gene mutation. Pancreatology 2015; 15:194-6. [PMID: 25464936]
- Mao Y, Sang X, Liang N, Yang H, Lu X, Yang Z, Du S, Xu Y, et al. Peripheral primitive neuroectodermal tumors arising in the pancreas: the first case report in Asia and a review of the 14 total reported cases in the world. Hepatobiliary Surg Nutr 2013; 2:51-60. [PMID: 24570916]
- Johnson PR. Gastroenteropancreatic neuroendocrine (carcinoid) tumors in children. Semin Pediatr Surg 2014; 23:91-5. [PMID: 24931354]
- Bülchmann G, Schuster T, Haas RJ, Joppich I. Primitive neuroectodermal tumor of the pancreas. An extremely rare tumor. Case report and review of the literature. Klin Padiatr 2000; 212:185-8. [PMID: 10994548]
- Movahedi-Lankarani S, Hruban RH, Westra WH, Klimstra DS. Primitive neuroectodermal tumors of the pancreas: a report of seven cases of a rare neoplasm. Am J Surg Pathol 2002; 26:1040-7. [PMID: 12170091]
- Petrosyan M, Franklin AL, Jackson HT, McGue S, Reyes CA, Kane TD. Solid pancreatic pseudopapillary tumor managed laparoscopically in adolescents: a case series and review of the literature. J Laparoendosc Adv Surg Tech A 2014; 24:440-4. [PMID: 24746104]

