Keywords
Pancreatic Neoplasms; Pharmacogenetics; Therapeutics
Abbreviation
5'-DFUR: 5'-deoxy-5- fluorouridine; ABCB1: ATP-binding cassette, sub-family B (MDR/TAP), member 1; ABCC2: ATP-binding cassette, sub-family C (CFTR/MRP), member 2; ABCG2: ATPbinding cassette, sub-family G (WHITE), member 2; AKT2: v-akt murine thymoma viral oncogene homolog 2; BAX: BCL2- associated X protein; BCL2: B-cell CLL/lymphoma 2; BCL2L1 (also know as Bcl-xl): BCL2-like 1; BRAF: v-raf murine sarcoma viral oncogene homolog B1; BH3: BCL2 homology 3; BNIP3: BCL2/adenovirus E1B 19kDa interacting protein 3; BRCA1: breast cancer 1, early onset; BRCA2: breast cancer 2, early onset; CDA: cytidine deaminase; CDKN2A (also known as p16INK4): cyclin-dependent kinase inhibitor 2A; CI-1040: 2-(2-chloro-4-iodophenylamino)- N-cyclopropylmethoxy-3,4-difluorobenzamide; c-SRC: v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog; CTP: cytidine triphosphate; dCK: deoxycytidine kinase; dCMP: deoxycytidine monophosphate; dCTP: deoxycytidine triphosphate; dFdC: difluorodeoxycytidine; dFdCDP: difluorodeoxycytidine diphosphate; dFdCTP: difluorodeoxycytidine 5'-triphosphate; DPD: dehydropyrimidine dehydrogenase; dUMP: deoxyuridylate; dUTP: deoxyuridine triphosphate; EGFR (also known as ErbB1): epidermal growth factor receptor; ERBB2 (also known as HER-2/neu): v-erb-b2 erythroblastic leukemia viral oncogene homolog 2; ERCC1: excision repair cross-complementing rodent repair deficiency, complementation group 1; ERK: elk-related tyrosine kinase; FAK: focal adhesion kinase; FdUMP: fluorodeoxyuridylate; FdUTP: fluoro-deoxyuridine triphosphate; FUTP: fluorouridine triphosphate; GSTM1: glutathione S-transferase M1; GSTP1: glutathione S-transferase pi; GSTT1: glutathione Stransferase theta 1; hCNT: concentrative nucleoside transporters (humans); hENT: equilibrative nucleoside transporters (humans); I-DNA: type 1 DNA topoisomerase; MAP2K2 (also known as MEK2): mitogenactivated protein kinase kinase 2; MEK1: mitogen-activated protein kinase kinase 1; MAP2K4 (also known as SEK1): mitogenactivated protein kinase kinase 4; MLH1: mutL homolog 1, colon cancer, nonpolyposis type 2; MMR: mismatch repair; MTHFR: methylene tetrahydrofolate reductase; MYB: v-myb myeloblastosis viral oncogene homolog; NCOA3 (also known as AIB1): nuclear receptor coactivator 3; OPRT: orotate phosphorylase transferase; PKI-166: 4- phenethylamino-6-(yderoxyl)phenyl-7H-pyrrolo (2,3-d)pyrimidine; RB1: retinoblastoma 1; RR: ribonucleotide reductase; RRM: ribonucleotide reductase M; siRNA: small interfering RNA; SMAD4 (also known as DPC4): SMAD family member 4; SN-38: 7- ethyl-l0-hydroxycamptothecin; SN-38G: SN- 38 glucuronide; SNP: single nucleotide polymorphisms; STK11 (also known as LKB1): serine/threonine kinase 11; TGFBR2: transforming growth factor, beta receptor II; TP: thymidine phosphorylase; TP53: tumor protein p53; TS: thymidylate synthase; TSER: TS enhancer region; UDP: uridine diphosphate; UGT1A1: UDP glucuronosyltransferase 1 family, polypeptide A1; UMPK: uridine monophosphate kinase; XRCC1: Xray repair complementing defective repair in Chinese hamster cells 1
INTRODUCTION
Each year approximately 30, 000 Americans are being diagnosed with pancreatic cancer and nearly all succumb to their disease. It is one of the most challenging solid tumors due to its aggressiveness, debilitating diseaserelated symptoms, and chemo-resistant biology. At present time, single agent gemcitabine is regarded as the standard of care for advanced pancreatic cancer after a randomized trial showing it superior clinical benefit response over 5-FU. A numerous randomized trials testing gemcitabine doublets failed to show survival benefit by addition of 2nd agent except for one UK study suggesting improved survival with addition of capecitabine [1]. Platinum based agents, topoisomerase inhibitors, taxanes, and novel biological agents have been tried as single agent and as combination with gemcitabine without significant improvement in the outcomes. Our efforts to optimize currently available treatment options and to develop novel agents for pancreatic cancer must start from better understanding of its biology.
Extensive research in the past two decades has revealed that pancreatic cancer is a genetic disease which is associated with various forms of cancer associated genetic alterations. Oncogenes, tumor suppressor genes, and DNA mismatch genes are known to be altered via various modes leading to cancer development, progression, chemoresistance, and in some cases chemosensitivities of pancreatic cancer [1, 2, 3].
Basics of Pancreatic Cancer Genetics
Oncogenes, when activated, can promote cancer development and progression. K-ras gene is aberrantly activated in up to 90% of pancreatic cancer [2] (Table 1). V-akt murine thymoma viral oncogene homolog 2 (AKT2) gene and nuclear receptor coactivator 3 (NCOA3; also known as AIB1) gene mutations have also been associated with pancreatic cancer.

Tumor suppressor genes are inactivated by homozygous deletion, combination of mutation of one copy and loss of the other copy, and aberrant hypermethylation of gene promotor causing the silencing of the gene. There are multiple tumor suppressor genes associated with pancreatic cancer and they are mitogen-activated protein kinase kinase 4 (MAP2K4; also known as SEK1), serine/ threonine kinase 11 (STK11; also known as LKB1), transforming growth factor, beta receptor II (TGFBR2), tumor protein p53 (TP53) and cyclin-dependent kinase inhibitor 2A (CDKN2A; also known as p16INK4). DNA mismatch repair genes codes for the proteins that correct errors made randomly during DNA replications, and inactivation of these genes has been associated with various cancers including pancreatic cancers.
Identification and understanding of these carcinogenic gene alterations is the base upon which we can overcome drug resistance and develop novel treatment approaches. Therapeutic interventions that are geared to reverse this process are actively being investigated.
CYTOTOXIC AGENTS (Table 2)
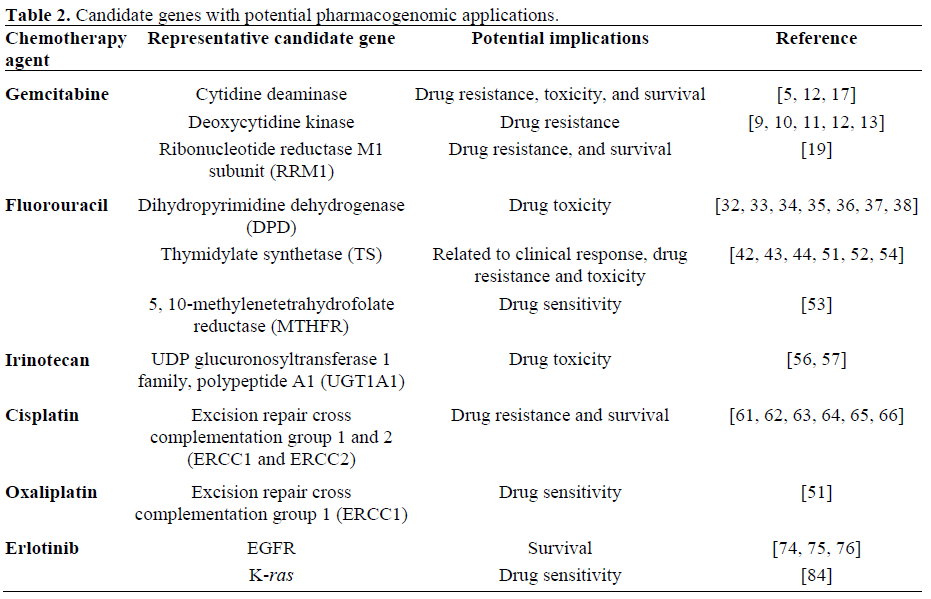
Gemcitabine
5-FU has been the primary agent for palliative treatment of advance pancreatic cancer until when a randomized trial in mid 1990’s showed gemcitabine’s superiority over 5-FU in terms of clinical benefits response (24% versus 5%) and 1-year survival (18% versus 2%; P=0.0022). Since then gemcitabine has been the backbone agent in treatment of pancreatic cancer as single agent as well as combinations with other agents. Gemcitabine is relatively well tolerated with common side effects of grade 1 or 2 myelosuppression, fatigue, and gastrointestinal symptoms.
Mechanism of Action
Gemcitabine (2’,2’-difluorodeoxycytidine) is a fluorine-substituted deoxycytidine analog. It requires intracellular activation by deoxycytidine kinase to the monophosphate form with eventual metabolism to the cytotoxic nucleotide metabolite difluorodeoxycytidine 5'-triphosphate (dFdCTP). Antitumor activity of gemcitabine is determined by a balance between intracellular activation and degradation and the formation of cytotoxic triphosphate metabolites. Incorporation of dFdCTP metabolite inhibits several DNA polymerases which in turn, interferes with DNA chain elongation, DNA synthesis, and DNA repair. Difluorodeoxycytidine diphosphate (dFdCDP) metabolites inhibit the enzyme ribonucleotide reductase, resulting in decreased levels of essential deoxyribonucleotides for DNA synthesis and function. Incorporation into RNA resulting in alterations in RNA processing and mRNA translation.
Machanisms of Drug Resistance
Currently understood mechanisms of gemcitabine resistance are following: i. decreased expression of rate limiting enzyme deoxycytidine kinase leading to subsequent decreased activation of gemcitabine;
ii. increased breakdown of drug by catabolic enzymes cytidine deaminase and deoxycytidine monophosphate (dCMP) deaminase; iii. decreased nucleoside transport of drug into cells;
iv. increased concentration of competing physiologic nucleotide deoxycytidine triphosphate (dCTP), through increased expression of cytidine triphosphate (CTP) synthetase.
Pharmacogenomics
Equilibrative nucleoside transporters (hENT), and concentrative nucleoside transporters (hCNT) are proteins that transport gemcitabine to intracelluar targets. There are over 58 hCNT single nucleotide polymorphisms (SNPs) identified with 3 SNPs with probable functional impact. In vitro studies revealed that gemcitabine is mostly transported to intracellular targets via hENT1 transporters [4]. At least 24 hENT SNPs are also being identified however, the functional impact of these SNPs are being investigated [5, 6]. There are in vitro data showing that gemcitabine sensitivity may be significantly altered by inhibiting these transporter proteins [7]. There are studies also suggesting that high expression of hENT1 is associated with survival benefits in pancreatic cancer patients [8].
Deoxycytidine kinase (dCK) is viewed as a rate-limiting enzyme in activating gemcitabine after cellular uptake by transporters. Decreased expression of this enzyme has been reported to be one of the mechanisms of gemcitabine resistance. Correlation between pancreatic cancer's sensitivity to gemcitabine and dCK has been well described [9, 10, 11]. dCK enzyme activity modulating agents are being investigated to design a combination regimen that will optimize gemcitabine's antitumor activity [12, 13].
Studies suggested that infusion of gemcitabine at a fixed dose rate of 10 mg/m2/min had pharmacodynamic advantages over 30 minute infusion by as the rate limiting enzyme dCK is known to be saturated at gemcitabine concentration of about 10 mg/m2/min [14]. Phase II trials showed that fixed dose rate gemcitabine can render a trend toward survival benefits [15]. However, the United States Intergroup study have failed to show any significant benefit with fixed dose rate while yielding higher incidence of toxicities [16].
Fixed dose rate infusion of gemcitabine in relation to dCK activity levels may be a complicated yet very important area of study to optimize gemcitabine therapy.
Gemcitabine is inactivated by cytidine deaminase (CDA) and high expression of this enzyme has been associated with gemcitabine resistance and shorter survival [12, 17]. CDA is a polymorphic enzyme and there is one variant allele (208G>A (3*, A70T)) that was identified in Japanese population (allele frequency of 0.037) that had functional impact of CDA. 3* allele appeared to change pharmacokinetic parameters and plasma CDA activities significantly leading to decreased clearance of gemcitabine and increased toxicity. More polymorphisms are being identified and their functional impact will need to be examined [5].
Resistance to gemcitabine is observed in ribonucleotide reductase (RR) M1 and M2 (RRM1 and RRM2) over-expressing lung cancer cells. RRM1 promoter allotypes 37CC-524TT influence overall survival and disease free survival in lung cancer [18]. The diphosphorylated form of gemcitabine inhibits RR and causes some of gemcitabine antitumor activity. M1 subunit of RR is reported to be an important target, and its over expression has been associated with gemcitabine resistance in non small cell lung cancer and pancreatic cancer [19]. Using microarray data, Nakahira et al. [19] have demonstrated that RRM1 gene was the most upregulated gene in the gemcitabine-resistant pancreatic cancer. Furthermore they were able to reduce gemcitabine resistance, in vitro, by RRM1 specific RNAi transfection and subsequent decreased RRM1 expression. They also reported statistically significant association between RRM1 over expression and poorer survival in pancreatic cancer patients [19]. These in vitro and clinical data strongly suggest that not only RRM1 expression level can be used as a predictor of gemcitabine resistance, but also it can be the target to modulate gemcitabine resistance.
Phosphatidylinositol 3-kinase/AKT and NFkappaB are antiapoptotic signal transduction pathways and liked to chemoresistance of pancreas cancer cells [20]. A reduction of phosphorylated protein kinase B/Akt correlated with the enhancement of gemcitabine induced apoptosis and antitumor activity [21]. NF-kappaB inhibition may enhance the activity of gemcitabine. A numerous lines of evidence suggest that NFkappaB plays a major role in the growth and chemoresistance of pancreatic cancer. Agents such as curcumin that down regulates NFkappaB and shown to potentiate antitumor activity of gemcitabine in pancreatic cancer cell lines are future area of clinical investigation [22, 23].
Nakano et al., using RT-PCR analysis, showed that the hENT1 x dCK / RRM1 x RRM2 genes expression level ratio correlated with gemcitabine resistance in pancreatic cancer cell lines indicating the complexity of pharmacogenomics [23].
V-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (c-SRC) and focal adhesion kinase (FAK) were also described as genes that are associated with gemcitabine resistance [24].
B-cell CLL/lymphoma 2 (BCL2) over expression is associated with increased in gemcitabine induced apoptosis, and expression of BCL2-associated X protein (BAX) increases sensitivity of pancreatic cancer cell lines to gem and 5-FU [25]. BCL2/adenovirus E1B 19kDa interacting protein 3 (BNIP3) is a member of the BCL2 homology 3 (BH3)-only subfamily of BCL2 family proteins that promote apoptosis. Akada et al. reported a strong association between BNIP3 and intrinsic resistance to gemcitabine in pancreatic cancer [26]. They also showed that by suppressing BNIP3 by small interfering RNA (siRNA), gemcitabine cytotoxicity was reduced in pancreatic cancer cell lines [26]. This suggests that BNIP3 could be a potential predictor of gemcitabine sensitivity and furthermore, is a possible target to increase gemcitabine sensitivity
Mutated p53 and BCL2-like 1 (BCL2L1; also know as Bcl-xl) have also been associated with gemcitabine resistance and they were directly involved in apoptosis [27, 28, 29]. Pharmacogenomics of gemcitabine discussed above is being investigated in a clinical trial setting [30].
FLUOROPYRIMIDINES
5-Fluorouracil (5-FU)
Mechanism of Action
5-FU is a fluoropyrimidine analog that inhibits the target enzyme thymidylate synthase (TS) by the 5-FU metabolite, fluorodeoxyuridylate (FdUMP). Inhibition of TS results in accumulation of deoxyuridylate (dUMP) which then gets misincorporated into DNA in the form of deoxyuridine triphosphate (dUTP) resulting in inhibition of DNA synthesis and function. Incorporation of the fluorouridine triphosphate (FUTP) into RNA results in alterations in RNA processing and translation, and incorporation of fluorodeoxyuridine triphosphate (FdUTP) into DNA results in inhibition of DNA synthesis and function.
Mechanisms of Drug Resistance
Mechanisms of drug resistance of 5-FU are following:
i. increased expression of TS;
ii. decreased levels of reduced folate substrate 5, 10-methylenetetrahydrofolate for TS reaction;
iii. decreased incorporation of 5-FU into RNA and DNA;
iv. increased salvage of physiologic nucleosides including thymidine;
v. decreased expression of mismatch repair enzymes;
vi. increased expression of dihydropyrimidine dehydrogenate;
vii. alterations in TS with decreased binding affinity of enzyme for FdUMP.
Capecitabine
Capecitabine is a pro-drug that is converted to 5'-deoxy-5-fluorouridine (5'-DFUR) in the liver and tumor tissue in three sequential enzymatic reactions. The final requisite enzyme thymidine phosphorylase (TP) is present at higher levels in tumor compare to normal tissue, thereby having selective antitumor activity. It is the first oral fluoropyrimidine-based anticancer drug that the American FDA has approved as first-line chemotherapy for the indication of metastatic colon cancer [31].
Pharmacogenomics
Earlier pharmacogenomic studies demonstrated that response to 5-FU depends on intra-tumor levels of dehydropyrimidine dehydrogenase (DPD) and TS. High expression of DPD increases 5-FU catabolism (leading to inactivation and elimination) while low levels lead to decreased 5-FU clearance and increased anabolism (cytotoxic pathway) [32, 33, 34, 35, 36, 37, 38]. Similarly, the importance of TS as a target of 5-FU has been underscored by studies demonstrating that incomplete inhibition of TS in the tumor results in reduced chemotherapy effect [39, 40, 41]. Role of increased tumor levels of DPD and/or TS as a mechanism of resistance has been demonstrated in patients treated with 5- FU [42, 43, 44]. Reports also show that high TP in stomach cancer and colon cancer is associated with a bad prognosis [45, 46].
Studies also demonstrated that intra-tumor levels of TP and DPD are good indicators of tumor response to capecitabine [47, 48, 49]. TP is an enzyme that catalyzes the mutual transformation of the pyrimidine nucleosides thymidine and thymine in nucleic acid metabolism. TP is also an enzyme that converts the 5-FU-based anticancer drugs, 5'- DFUR and its derivative, capecitabine, into 5- FU, and it is therefore a limiting factor of the anti-tumor effects of these anticancer drugs. Increased TP results in higher intra-tumor 5- FU level thereby enhancing anti-tumor effect of capecitabine. This preferential activation of capecitabine to 5-FU was demonstrated in colorectal tumor by Schüller J et al. [49]. Tsukamoto et al. presented in vitro data suggesting that high DPD expression results in lower intra-tumor 5-FU levels through increased degradation [48].
An in vitro study suggested the importance of the ratio of TP and DPD (TP:DPD) in predicting activity of capecitabine against cancer cells. Ishikawa et al. showed that capecitabine can be effective in tumors expressing low TP if DPD expression was low as well [47]. Conversely, capecitabine was not as effective as it was expected to be in tumors with high TP levels if tumors had high DPD levels [47]. Other recent studies suggested correlation between high TP:DPD and anti-cancer activity of 5’-DFUR in human cancer xenograft models [50]. A recent study using 5’-DFUR as adjuvant chemotherapy in colorectal cancer showed that patients with high TP:DPD had better disease-free survival which supported the hypothesis that 5-FU is better activated with high TP levels and degraded to a lesser degree with low DPD levels thereby giving cancer cells maximum exposure its anticancer activity [50, 51].
Genetic polymorphisms of enzymes involved in the fluoropyrimidines have been described. Exact functional impact of polymorphisms of uridine monophosphate kinase (UMPK) and orotate phosphorylase transferase (OPRT) are unknown as of yet.
Polymorphism of TS is better described. At least 5 genotypes have been described. They are characterized by numbers of a 28-bp tandem repeat sequence in the DNA promoter TS enhancer region (TSER). The number of tandem repeats vary from 2 copies (2R) to 9 copies (9R). TS expression level seems to correlate with the number of tandem repeats in in vitro study. Studies have shown that colorectal cancer patients with homozygous 2R TS genotypes have better clinical response with treatment with 5-FU compare to patients with 3R TS genotypes. In terms of toxicities, patients with 3R TS genotypes suffered less 5-FU toxicities probably due to the fact that higher TS level leading to less efficient TS inhibition and decreased cell death in cancer cells as well as normal cells [51, 52]. So far, at least two methylene tetrahydrofolate reductase (MTHFR) polymorphisms have been identified and pre-clinical and clinical data suggested their role as a modulators of fluoropyrimidine sensitivity [53]. Okumura et al. reported that TS mRNA expression level may be a predictor of chemosensitivity to 5- FU in colorectal cancer [54].
5-FU catabolic enzymes and their polymorphisms can have significant impact on 5-FU toxicities. Variability in the expression of DPD is a cause of variable 5- FU response and toxicity. Aberrant methylation of DPD promoter region can down regulate DPD activity and consequently increases risk of 5-FU toxicity in cancer patients [55].
Over 40 different polymorphisms have been identified and 17 mutations were found in patients with severe 5-FU toxicities. DPD*2A is the most commonly noted polymorphism with well established association with decreased DPD activity and 5-FU toxicity [52].
Other SNPs and haplotypes are being identified and their functional impact is being examined. They will offer potential targets to modulate response and tolerance of 5-FU in pancreatic cancer.
The Pharmacogenomic Rationale for a Combination of Gemcitabine and 5-FU in Pancreatic Cancer
A numerous studies examining synergy between 5-FU and gemcitabine revealed that there is no benefit of adding 5-FU to gemcitabine. However, there are phase II and phase III data suggesting benefit of gemcitabine plus capecitabine especially in patients with good performance status. A better patient selection based upon pharmacogenomics of both agents may improve the outcome of patients treated with this doublet chemotherapy.
Irinotecan
Mechanisms of Action
Irinotecan is a semisynthetic derivative of camptothecin, an alkaloid extract from the Camptotheca acuminata tree. It is activated by the carboxylesterase enzyme to its active form 7-ethyl-l0-hydroxycamptothecin (SN- 38). SN-38 binds to and stabilizes the type 1 DNA topoisomerase (I-DNA) complex and prevents the religation of DNA after it has been cleaved by topoisomerase I. The collision between this stable complex ("cleavable complex") and the advancing replication fork results in double-strand DNA breaks and cellular death.
Mechanisms of Drug Resistance
i. Decreased expression of topoisomerase I and mutations that decreases affinity for the drug are proposed mechanisms of irinotecan resistance;
ii. increased expression of the multidrugresistant phenotype with over expression of P170 glycoprotein and subsequent enhanced efflux of drug and decreased intracellular accumulation of drug is also thought to be associated with irinotecan resistance;
iii. decreased formation of the cytotoxic metabolite SN-38 through decreased activity and or expression of the carboxylase enzyme; iv. decreased accumulation of drug into cells by mechanisms not well identified.
P harmacogenomics
The final step of irinotecan metabolism is the inactivation of SN-38 by glucuronidation to SN-38 glucuronide (SN-38G). This step is mediated by uridine diphosphate (UDP) glucuronosyltransferase 1 family, polypeptide A1 (UGT1A1). Recent studies suggested that polymorphisms in the UGT1A1 gene may have an impact on irinotecan toxicities [56]. A variant allele, UGT1A1*28, was associated with irinotecan toxicity by causing reduced conversion of SN-38 in clinical trials. More studies to validate this genotype-phenotype relationship will help us optimize our use of irinotecan based upon the genetic profile in the future [56, 57].
Up to 50% of patients with UGT1A1*28 allele may experience severe irinotecan toxicity, we need to establish genotypeadjusted dosages of irinotecan. genotypeadjusted dosages of irinotecan are on going [57]. ATP-binding cassette, sub-family B (MDR/TAP), member 1 (ABCB1; a Pglycoprotein), and ATP-binding cassette, subfamily C (CFTR/MRP), member 2 (ABCC2; a multidrug resistance-associated protein) and ATP-binding cassette, sub-family G (WHITE), member 2 (ABCG2; a breast cancerresistance protein) are transmembrane proteins that facilitate the cellular efflux of irinotecan and its metabolites. SNPs of ABCB1 gene have been associated with increased exposure to irinotecan and SN-38 and certain haplotypes were associated with decreased renal clearance of irinotecan and SN-38. ABCC2 is the primary transporter involved in hepatobiliary secretion of irinotecan, SN-38, and SN-38G. A ABCC2*2 haplotype, recently, was associated with less irinotecan related toxicities, once again suggesting the possibility of individualizing treatment based upon genetic profiles [57].
The modulation of carboxylesterase expression may impact irinotecan sensitivity. Only 10% of irinotecan is converted to SN-38 by carboxylesterase enzymes [58, 59]. There has been a study that suggested that inducing over expression of carboxylesterase encoding gene in tumor cells, the efficacy of irinotecan can be improved [59].
In general, knowledge of target gene is still lacking. However, the significant achievement of irinotecan pharmacogenomics is a positive example of individualized chemotherapy.
The Pharmacogenomic Rationale for Combination of Gemcitabine and Irinotecan in Pancreatic Cancer
Preclinical study in cancer cell lines showed more than additive effect when SN-38 and gemcitabine suggesting synergy between these two agents [60]. However despite promising early phase trial results, phase III trials revealed no added benefit of irinotecan to gemcitabine alone. At present time, there is poor clinical or pharmacogenomic-based rationale in this combining irinotecan with gemcitabine in treatment of pancreatic cancer.
PLATINUM-BASED AGENTS IN PANCREATIC CANCER TREATMENT
Platinum based agents such as cisplatin and oxaliplatin have been used in treatment of pancreatic cancer in combination with gemcitabine with modest benefit evidenced in clinical trials.
Cisplatin
Mechanism of Action
Cisplatin covalently binds to DNA to produce DNA adducts that results in inhibition of DNA synthesis and function as well as transcription.
Mechanism of Drug Resistance
i. Increased DNA repair enzyme activity and deficiency in mismatch repair (MMR) enzymes;
ii. increased inactivation by thiol-containing proteins such as glutathione and glutathionerelated enzymes;
iii. alteration in cellular transport resulting in decreased drug accumulation.
Pharmacogenomics
Cisplatin resistance is associated with overexpression of excision repair crosscomplementing rodent repair deficiency, complementation group 1 (ERCC1) gene in ovarian cancer patients. A numerous preclinical and clinical trials have suggested that non small cell cancer patients with low ERCC1 expression levels have better outcome with cisplatin based regimen compared to patients with high ERCC1 expression [61, 62, 63]. Polymorphisms in ERCC1 were associated with prognosis and cisplatin resistance. A study observed that advanced lung cancer patients with one of common SNPs of ERCC1, C8092A, had significantly shorter median survival compared to patients without the variant alleles [64].
Bellmunt et al. suggested ERCC1 expression as a prognostic and predictive tool in treatment of bladder cancer. This study reported significantly prolonged survival of patients who were treated with cisplatin-based chemotherapy, hence supporting hypothesis that enhanced DNA repair decreases the benefit of platinum-based chemotherapy [65]. Role of breast cancer 1, early onset (BRCA1) gene in platinum resistance also has been suggested in literatures as well. Deficiency of BRCA1, which has an important role in DNA repair, is associated with resistance to platinum based regimens [66].
Pharmacogenomic Rationale for Gemcitabine and Cisplatin
Several in vitro studies using different cancer cell lines have suggested synergy between gemcitabine and cisplatin. The mechanism of this synergy is thought to be related to the incorporation of gemcitabine’s active metabolite, difluorodeoxycytidine (dFdC), into DNA that suppresses nuclear excision repair and subsequent increase of cisplatin induced DNA adduct formation [67, 68].
Oxaliplatin
Mechanism of Action
Oxaliplatin is a third generation platinum compound that binds to DNA to produce DNA adducts resulting in DNA synthesis inhibition as well as inhibition of transcription.
Mechanism of Resistance
The mechanisms of oxaliplatin resistance are similar to those of cisplatin.
Pharmacogenomics
In vitro studies show an antitumor activity of oxaliplatin against pancreatic cancer cell lines [51, 69].
Clinical data show correlation between TS/ERCC1 expression level and overall survival in colorectal cancer patients who were treated with 5-FU/oxaliplatin suggesting the role of ERCC1 in oxaliplatin drug sensitivity in similar mechanism as described above in review of cisplatin drug resistance [51].
Various polymorphisms of glutathione Stransferase pi (GSTP1), Xeroderma pigmentosum group D protein, glutathione Stransferase M1 (GSTM1), glutathione Stransferase theta 1 (GSTT1), ERCC1 T19007C, X-ray repair complementing defective repair in Chinese hamster cells 1 (XRCC1), and epidermal growth factor receptor (EGFR; also known as ErbB1) were associated with clinical outcome of oxaliplatin treatment [51].
Pharmacogenomic Rationale for Gemcitabine and Oxaliplatin Combination
The rational for the use of a combination regimen of gemcitabine plus oxaliplatin stems from various preclinical data. The exposure to gemcitabine 24 hours prior to oxaliplatin showed synergistic effects in antitumor effects in colorectal and leukemia cell lines [70].
The precise mechanisms of this synergy is poorly described, however the incorporation of gemcitabine into DNA subsequently increasing DNA adduct is thought to be the most likely mechanism [71].
NOVEL APPROACHES: TARGETED AGENTS
EGFR is over expressed by about 80% pancreatic cancer. It has been associated with poorer survival [72], and it has become an important target for novel anticancer therapy.
Erlotinib
Erlotinib is a small molecule tyrosine kinas inhibitor targeting the intracellular domain of EGFR and compete with ATP for binding to the kinase domain, thereby impeding downstream signaling. Erlotinib has shown modest activity as a single agent in pancreatic cancer, and it is currently approved by FDA in combination with gemcitabine in metastatic pancreatic cancer. Skin rash was strongly correlated with better treatment outcome, and a recent data presented at the 2007 ASCO Annual Meeting suggested that the absence of K-ras mutation was associated with better treatment outcome with erlotinib [72].
Cetuximab
Cetuximab is a recombinant humanized monoclonal antibody directed against the EGFR. Inhibition of the EGFR signaling pathway results in inhibition of critical mitogenic and anti-apoptotic signals involved in proliferation, growth, invasion/metastasis, angiogenesis, and it also enhances the response to chemotherapy and radiation therapy. Decreased expression of EGFR and mutation in EGFR are thought to be the mechanisms of drug resistance. Despite promising results from phase II studies, recently presented Phase III trial showed no additive benefit of cetuximab to gemcitabine when this combination regimen was compared to gemcitabine alone [73].
Pharmacogenomics of Anti EGFR Therapy
In lung cancer, patients with EGFR mutations had better outcomes with use of erlotinib than patients without the mutation. However, in studies using erlotinib in other cancer types, EGFR mutation status was poorly studied. The modest benefit of erlotinib plus gemcitabine versus gemcitabine alone in a phase III trial warrants more pharmacogenomic investigation. Studies report mere 1.5 to 3.6% frequency of EGFR mutations in pancreatic cancers which is significantly lower that other cancer types. A recent study of Lee et al. [74] also observed that the presence of EGFR mutation or increased gene copy number did not significantly influence the survival of pancreatic cancer patients; this is in contrast to well accepted data [75, 76]. This study also suggest that in pancreatic cancer, EGFR mutation may not be predictive of sensitivity to EGFR tyrosine kinase inhibitors [74]. A study by Tzeng et al. characterized EGFR mutations in pancreatic adenocarcinomas by using 9 pancreatic carcinoma cell lines and 31 specimens from pancreatic cancer patients, and they observed that EGFR tyrosine kinase domain is highly conserved in pancreatic cancer [77]. This observation may be the explanation of disappointing outcome of anti-EGFR therapy in pancreatic cancer patients. There needs to be more clinical investigations of predictive values of the EGFR mutation and gene copy number on erlotinib or gefitinib response in pancreatic cancer patients in the future.
Various EGFR modulations with cetuximab results in inhibition of DNA repair as well as inhibition of tumor angiogenesis. Recent data by Sung et al. [78] suggest that cetuximab may increase sensitivity to DNA damaging chemotherapy agents.
Matuzumab is another monoclonal antibody against EGFR that has shown activity in combination with gemcitabine in an early phase clinical trial [79]. 4-phenethylamino-6- (yderoxyl)phenyl-7H-pyrrolo(2,3-d)pyrimidine (PKI-166), a novel dual inhibitor of EGFR and v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERBB2; also known as HER-2/neu), has been developed and has been promising in preclinical and early clinical trials [80]. Lapatinib is the other dual tyrosine kinase inhibitors tested more extensively in breast cancer patients [81]. Trials in pancreatic cancer are underway
ERBB2 (also known as HER-2/neu)
ERBB2 is an epithelial tumor oncogenes amplified and over expressed in breast, ovary, colon, lung, salivary gland and pancreatic carcinomas. Unlike in breast cancer, significance of ERBB2 over expression is unclear in pancreatic cancer
Despite promising preclinical data showing activity against pancreatic cancer cell lines, orthotopic mouse model, and early clinical trials, Safran et al. showed that trastuzumab and gemcitabine combination is not superior to gemcitabine alone in a clinical trial [82].
Anti-VEGF Agents
Over-expression of VEGF is associated with early recurrence after surgery and shorter survival [83]. Based on promising preclinical data, bevacizumab, a recombinant humanized anti-VEGF monoclonal antibody, was tested in pancreatic cancer patients. Kindler et al., in the 2007 ASCO Annual Meeting, presented data showing that bevacizumab and gemcitabine combination is not superior to gemcitabine alone in metastatic pancreatic cancer patients [73].
Sorafinib is a small molecule multi tyrosine kinase inhibitor. The combination of gemcitabine and sorafinib failed to render superiority to gemcitabine alone.
Pharmacogenomics
Several polymorphisms of VEGF gene are identified. While there were association of this variant alleles and prognosis of ovarian and breast cancer, the implication of these SNPs in relation to sensitivity to anti-VEGF treatment in poorly studied. A better understanding of target mutation status and biological consequences will benefit monoclonal antibody development and may guide clinical development and use of these innovative agents
Concomitant Inhibition of EGFR and VEGF
AEE788 is a novel agent that inhibits both EGFR and VEGF receptor tyrosine kinases. In vitro studies confirmed its anti-tumor activity. AEE788 in combination with gemcitabine resulted in inhibition of tumor growth, enhancement of apoptotic activity, reduction of microvessel density, and improved survival in orthotopic pancreatic cancer xenograft model [83].
K-ras as a Target
K-ras oncogene is activated by point mutations in up to 90% of pancreatic cancer cases with a “signature” localization in codon 12. K-ras mutation in pancreatic cancer is known to develop during the early phase of pancreatic cancer carcinogenesis. Studies suggested that K-ras mutation is associated with poor clinical outcomes. K-ras mutation which can be detected with various means including peripheral blood, has a potential to become a tool for early diagnosis of pancreatic cancer and in order to select patients who will benefit from erlotinib as patients with K-ras mutation are less likely to respond to erlotinib than patients without the mutation [84].
Farnesyl transferase is the enzyme catalyzing the synthesis of ras-protein. Several agents that inhibits this enzyme were developed and showed promising activities in preclinical studies. Tipifarnib was tested as a monotherapy as well as a combination therapy with gemcitabine, and the results were disappointing. Tipifarnib in phase III trial did not prolong survival in advanced pancreatic cancer compared with gemcitabine alone [85].
Other Agents
Matrix Metalloproteases
The matrix metalloproteases play an important role in the growth, migration, invasion, metastasis formation, angiogenesis in cancer. Marimastat showed promising results in pancreatic cancer in early studies, but in phase III double blind study, there was no benefit of adding marimastat [86].
Cyclooxygenase-2 Inhibition
Cyclooxygenase is an enzyme that is known to contribute to the growth of pancreatic cancer. Preclinical data showed marked inhibition of cell proliferation and induction of apoptosis by treatment with a non steroidal anti inflammatory drugs. Celecoxib, a COX-2 inhibitor, significantly enhanced anti tumor activity of chemoradiation in locally advanced pancreatic cancer patients in one study. Other studies failed to show benefit of celecoxib in combination with other chemotherapy agents. In addition, COX-2 inhibitors add significant toxicities, and there are data suggesting that COX-2 inhibitors may increase VEGF production and angiogenesis.
Signal Transduction Inhibitors and Cell Cycle Regulators
2-(2-chloro-4-iodophenylamino)-N-cyclopropylmethoxy- 3,4-difluorobenzamide (CI-1040) is an inhibitor of mitogen-activated protein kinase kinase 1 (MEK1) and mitogenactivated protein kinase kinase 2 (MAP2K2; also known as MEK2) thereby blocking the phosphorylation of elk-related tyrosine kinase (ERK). A Phase II study showed modest activity against advanced pancreatic cancer with acceptable toxicities. NF-kappa B is over expressed in pancreatic cancer and it has become a target of therapy. Curcumin has antiproliferative activity in pancreatic cancer cell lines. Liposome-encapsulated curcumin decreases tumor growth and neoangiogenesis in preclinical setting. An antisense agent against H-ras (ISIS-2503) has been tried in phase II study in combination with gemcitabine and failed to show its superiority to gemcitabine alone.
Endocrine Manipulation
Anti gastrin therapy and somatostatin analogs have shown encouraging result in pancreatic cancer in preclinical studies. However, clinical trials using hormonal therapies have been largely discouraging.
CONCLUSION AND FUTURE DIRECTION
Therapies currently used in treatment of pancreatic cancer have shown largely disappointing efficacies. New strategies need to be placed in order to move forward. The completion of human genome project has increased interests in the field of pharmacogenomics, and advancement of biotechnology is making application of genomic data more affordable and practical. We are gradually moving toward realizing individualized cancer treatment with pharmacogenomic data that can improve the outcomes of currently available treatments by better patient selection and novel drug administration schema as well as intelligent combinations. However, there still lies great challenges in front of us in practical use of pharmacogenomics data.
Most importantly, we must invest our resources in technological development to identify more important candidate genes and to make those assays available and affordable. With decreasing cost of genotyping, clinical investigators must make concerted efforts to incorporate pharmacogenomics endpoints to large cooperative clinical trials in order to approach required power to test significance of candidate genes. p16INK4): cyclin-dependent kinase inhibitor 2A; CI-1040: 2-(2-chloro-4-iodophenylamino)- N-cyclopropylmethoxy-3,4-difluorobenzamide; c-SRC: v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog; CTP: cytidine triphosphate; dCK: deoxycytidine kinase; dCMP: deoxycytidine monophosphate; dCTP: deoxycytidine triphosphate; dFdC: difluorodeoxycytidine; dFdCDP: difluorodeoxycytidine diphosphate; dFdCTP: difluorodeoxycytidine 5'-triphosphate; DPD: dehydropyrimidine dehydrogenase; dUMP: deoxyuridylate; dUTP: deoxyuridine triphosphate; EGFR (also known as ErbB1): epidermal growth factor receptor; ERBB2 (also known as HER-2/neu): v-erb-b2 erythroblastic leukemia viral oncogene homolog 2; ERCC1: excision repair cross-complementing rodent repair deficiency, complementation group 1; ERK: elk-related tyrosine kinase; FAK: focal adhesion kinase; FdUMP: fluorodeoxyuridylate; FdUTP: fluoro-deoxyuridine triphosphate; FUTP: fluorouridine triphosphate; GSTM1: glutathione S-transferase M1; GSTP1: glutathione S-transferase pi; GSTT1: glutathione Stransferase theta 1; hCNT: concentrative nucleoside transporters (humans); hENT: equilibrative nucleoside transporters (humans); I-DNA: type 1 DNA topoisomerase; MAP2K2 (also known as MEK2): mitogenactivated protein kinase kinase 2; MEK1: mitogen-activated protein kinase kinase 1; MAP2K4 (also known as SEK1): mitogenactivated protein kinase kinase 4; MLH1: mutL homolog 1, colon cancer, nonpolyposis type 2; MMR: mismatch repair; MTHFR: methylene tetrahydrofolate reductase; MYB: v-myb myeloblastosis viral oncogene homolog; NCOA3 (also known as AIB1): nuclear receptor coactivator 3; OPRT: orotate phosphorylase transferase; PKI-166: 4- phenethylamino-6-(yderoxyl)phenyl-7H-pyrrolo (2,3-d)pyrimidine; RB1: retinoblastoma 1; RR: ribonucleotide reductase; RRM: ribonucleotide reductase M; siRNA: small interfering RNA; SMAD4 (also known as DPC4): SMAD family member 4; SN-38: 7- ethyl-l0-hydroxycamptothecin; SN-38G: SN- 38 glucuronide; SNP: single nucleotide polymorphisms; STK11 (also known as LKB1): serine/threonine kinase 11; TGFBR2: transforming growth factor, beta receptor II; TP: thymidine phosphorylase; TP53: tumor protein p53; TS: thymidylate synthase; TSER: TS enhancer region; UDP: uridine diphosphate; UGT1A1: UDP glucuronosyltransferase 1 family, polypeptide A1; UMPK: uridine monophosphate kinase; XRCC1: Xray repair complementing defective repair in Chinese hamster cells 1
Conflict of interest
The authors have no potential conflicts of interest
References
- Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg 2007; 14:224- 32. [PMID 17520196]
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988; 53:549-54. [PMID 2453289]
- Hruban RH, Fukushima N. Pancreatic adenocarcinoma: update on the surgical pathology of carcinomas of ductal origin and PanINs. Mod Pathol 2007; 20(Suppl 1):S61-70. [PMID 17486053]
- García-Manteiga J, Molina-Arcas M, Casado FJ, Mazo A, Pastor-Anglada M. Nucleoside transporter profiles in human pancreatic cancer cells: role of hCNT1 in 2',2'-difluorodeoxycytidine- induced cytotoxicity. Clin Cancer Res 2003; 9:5000-8. [PMID 14581375]
- Abraham J, Earl HM, Pharoah PD, Caldas C. Pharmacogenetics of cancer chemotherapy. Biochim Biophys Acta 2006; 1766:168-83. [PMID 17141416]
- Efferth T, Volm M. Pharmacogenetics for individualized cancer chemotherapy. Pharmacol Ther 2005; 107:155-76. [PMID 15890408]
- Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 1998; 58:4349-57. [PMID 9766663]
- Spratlin J, Sangha R, Glubrecht D, Dabbagh L, Young JD, Dumontet C, et al. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res 2004; 10:6956-61. [PMID 15501974]
- Blackstock AW, Lightfoot H, Case LD, Tepper JE, Mukherji SK, Mitchell BS, et al. Tumor uptake and elimination of 2',2'-difluoro-2'-deoxycytidine (gemcitabine) after deoxycytidine kinase gene transfer: correlation with in vivo tumor response. Clin Cancer Res 2001; 7:3263-8. [PMID 11595723]
- Kroep JR, Loves WJ, van der Wilt CL, Alvarez E, Talianidis I, Boven E, et al. Pretreatment deoxycytidine kinase levels predict in vivo gemcitabine sensitivity. Mol Cancer Ther 2002; 1:371-6. [PMID 12477049]
- Giovannetti E, Mey V, Danesi R, Mosca I, Del Tacca M. Synergistic cytotoxicity and pharmacogenetics of gemcitabine and pemetrexed combination in pancreatic cancer cell lines. Clin Cancer Res 2004; 10:2936-43. [PMID 15131028]
- Spasokoukotskaja T, Sasvári-Székely M, Keszler G, Albertioni F, Eriksson S, Staub M. Treatment of normal and malignant cells with nucleoside analogues and etoposide enhances deoxycytidine kinase activity. Eur J Cancer 1999; 35:1862-7. [PMID 10674004]
- Giovannetti E, Mey V, Nannizzi S, Pasqualetti G, Del Tacca M, Danesi R. Pharmacogenetics of anticancer drug sensitivity in pancreatic cancer. Mol Cancer Ther 2006; 5:1387-95. [PMID 16818496]
- Abbruzzese JL. New applications of gemcitabine and future directions in the management of pancreatic cancer. Cancer 2002; 95:941-5. [PMID 12209675]
- Tempero M, Plunkett W, Ruiz Van Haperen V, Hainsworth J, Hochster H, Lenzi R, Abbruzzese J. Randomized phase II comparison of dose-intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol 2003; 21:3402-8. [PMID 12885837]
- Hochster HS. Newer approaches to gemcitabinebased therapy of pancreatic cancer: fixed-dose-rate infusion and novel agents. Int J Radiat Oncol Biol Phys 2003; 56:24-30. [PMID 12826248]
- Bengala C, Guarneri V, Giovannetti E, Lencioni M, Fontana E, Mey V, et al. Prolonged fixed dose rate infusion of gemcitabine with autologous haemopoietic support in advanced pancreatic adenocarcinoma. Br J Cancer 2005; 93:35-40. [PMID 15986033
- Rosell R, Felip E, Taron M, Majo J, Mendez P, Sanchez-Ronco M, et al. Gene expression as a predictive marker of outcome in stage IIB-IIIA-IIIB non-small cell lung cancer after induction gemcitabinebased chemotherapy followed by resectional surgery. Clin Cancer Res 2004; 10:4215s-9s. [PMID 15217961]
- Nakahira S, Nakamori S, Tsujie M, Takahashi Y, Okami J, Yoshioka S, et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer 2007; 120:1355-63. [PMID 17131328]
- Bondar VM, Sweeney-Gotsch B, Andreeff M, Mills GB, McConkey DJ. Inhibition of the phosphatidylinositol 3'-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol Cancer Ther 2002; 1:989-97. [PMID 12481421]
- Arlt A, Gehrz A, Müerköster S, Vorndamm J, Kruse ML, Fölsch UR, Schäfer H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003; 22:3243-51. [PMID 12761494]
- Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res 2007; 67:3853-61. [PMID 17440100]
- Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer 2007; 96:457-63. [PMID 17224927]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. siRNA directed against c-Src enhances pancreatic adenocarcinoma cell gemcitabine chemosensitivity. J Am Coll Surg 2004; 198:953-9. [PMID 15194078]
- Xu ZW, Friess H, Büchler MW, Solioz M. Overexpression of Bax sensitizes human pancreatic cancer cells to apoptosis induced by chemotherapeutic agents. Cancer Chemother Pharmacol 2002; 49:504-10. [PMID 12107556]
- Akada M, Crnogorac-Jurcevic T, Lattimore S, Mahon P, Lopes R, Sunamura M, et al. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin Cancer Res 2005; 11:3094-101. [PMID 15837765]
- Schniewind B, Christgen M, Kurdow R, Haye S, Kremer B, Kalthoff H, Ungefroren H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int J Cancer 2004; 109:182-8. [PMID 14750167]
- Galmarini CM, Clarke ML, Jordheim L, Santos CL, Cros E, Mackey JR, Dumontet C. Resistance to gemcitabine in a human follicular lymphoma cell line is due to partial deletion of the deoxycytidine kinase gene. BMC Pharmacol 2004; 4:8. [PMID 15157282]
- Shi X, Liu S, Kleeff J, Friess H, Büchler MW. Acquired resistance of pancreatic cancer cells towards 5-Fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology 2002; 62:354-62. [PMID 12138244]
- Javle MM, Okazaki T, Wolff RA, Varadhachary G, Ho L, Crane CH, et al. Combined effect of single nucleotide polymorphisms (SNPs) of gemcitabine metabolic genes on pancreatic cancer survival and drug toxicity. 2008 Gastrointestinal Cancers Symposium (abstract 260).
- Walko CM, Lindley C. Capecitabine: a review. Clin Ther 2005; 27:23-44. [PMID 15763604]
- Diasio RB, Johnson MR. The role of pharmacogenetics and pharmacogenomics in cancer chemotherapy with 5-fluorouracil. Pharmacology 2000; 61:199-203. [PMID 10971206]
- Diasio RB, Johnson MR. Dihydropyrimidine dehydrogenase: its role in 5-fluorouracil clinical toxicity and tumor resistance. Clin Cancer Res 1999; 5:2672-3. [PMID 10537327]
- Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res 2000; 6:1322-7. [PMID 10778957]
- Heggie GD, Sommadossi JP, Cross DS, Huster WJ, Diasio RB. Clinical pharmacokinetics of 5- fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res 1987; 47:2203-6. [PMID 3829006]
- Diasio RB, Beavers TL, Carpenter JT. Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity. J Clin Invest 1988; 81:47-51. [PMID 3335642]
- Huang CL, Yokomise H, Kobayashi S, Fukushima M, Hitomi S, Wada H. Intratumoral expression of thymidylate synthase and dihydropyrimidine dehydrogenase in non-small cell lung cancer patients treated with 5-FU-based chemotherapy. Int J Oncol 2000; 17:47-54. [PMID 10853017]
- Araki Y, Isomoto H, Shirouzu K. Dihydropyrimidine dehydrogenase activity and thymidylate synthase level are associated with response to 5-fluorouracil in human colorectal cancer. Kurume Med J 2001; 48:93-8. [PMID 11501504]
- Moran RG, Spears CP, Heidelberger C. Biochemical determinants of tumor sensitivity to 5- fluorouracil: ultrasensitive methods for the determination of 5-fluoro-2'-deoxyuridylate, 2'- deoxyuridylate, and thymidylate synthetase. Proc Natl Acad Sci U S A 1979; 76:1456-60. [PMID 108681]
- Yin MB, Zakrzewski SF, Hakala MT. Relationship of cellular folate cofactor pools to the activity of 5- fluorouracil. Mol Pharmacol 1983; 23:190-7. [PMID 6688119]
- Rustum YM, Cao S, Zhang Z. Rationale for treatment design: biochemical modulation of 5- fluorouracil by leucovorin. Cancer J Sci Am 1998; 4:12-8, [PMID 9467039]
- Hoque MO, Kawamata H, Nakashiro KI, Omotehara F, Shinagawa Y, Hino S, et al. Dihydropyrimidine dehydrogenase mRNA level correlates with the response to 5-fluorouracil-based chemo-immuno-radiation therapy in human oral squamous cell cancer. Int J Oncol 2001; 19:953-8. [PMID 11604993]
- Johnston PG, Lenz HJ, Leichman CG, Danenberg KD, Allegra CJ, Danenberg PV, Leichman L. Thymidylate synthase gene and protein expression correlate and are associated with response to 5- fluorouracil in human colorectal and gastric tumors. Cancer Res 1995; 55:1407-12. [PMID 7882343]
- Beck A, Etienne MC, Chéradame S, Fischel JL, Formento P, Renée N, Milano G. A role for dihydropyrimidine dehydrogenase and thymidylate synthase in tumour sensitivity to fluorouracil. Eur J Cancer 1994; 30:1517-22. [PMID 7833111]
- Nishida M, Hino A, Mori K, Matsumoto T, Yoshikubo T, Ishitsuka H. Preparation of anti-human thymidine phosphorylase monoclonal antibodies useful for detecting the enzyme levels in tumor tissues. Biol Pharm Bull 1996; 19:1407-11. [PMID 8951154]
- Mori K, Hasegawa M, Nishida M, Toma H, Fukuda M, Kubota T, et al. Expression levels of thymidine phosphorylase and dihydropyrimidine dehydrogenase in various human tumor tissues. Int J Oncol 2000; 17:33-8. [PMID 10853015]
- Ishikawa T, Sekiguchi F, Fukase Y, Sawada N, Ishitsuka H. Positive correlation between the efficacy of capecitabine and doxifluridine and the ratio of thymidine phosphorylase to dihydropyrimidine dehydrogenase activities in tumors in human cancer xenografts. Cancer Res 1998; 58:685-90. [PMID 9485021]
- Tsukamoto Y, Kato Y, Ura M, Horii I, Ishitsuka H, Kusuhara H, Sugiyama Y. A physiologically based pharmacokinetic analysis of capecitabine, a triple prodrug of 5-FU, in humans: the mechanism for tumorselective accumulation of 5-FU. Pharm Res 2001; 18:1190-202. [PMID 11587492]
- Schüller J, Cassidy J, Dumont E, Roos B, Durston S, Banken L, et al. Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother Pharmacol 2000; 45:291-7. [PMID 10755317]
- Nishimura G, Terada I, Kobayashi T, Ninomiya I, Kitagawa H, Fushida S, et al. Thymidine phosphorylase and dihydropyrimidine dehydrogenase levels in primary colorectal cancer show a relationship to clinical effects of 5'-deoxy-5-fluorouridine as adjuvant chemotherapy. Oncol Rep 2002; 9:479-82. [PMID 11956613]
- Lenz HJ. Pharmacogenomics and colorectal cancer. Adv Exp Med Biol 2006; 587:211-31. [PMID 17163168]
- Yong WP, Innocenti F, Ratain MJ. The role of pharmacogenetics in cancer therapeutics. Br J Clin Pharmacol 2006; 62:35-46. [PMID 16842377]
- Maring JG, Groen HJ, Wachters FM, Uges DR, de Vries EG. Genetic factors influencing pyrimidineantagonist chemotherapy. Pharmacogenomics J 2005; 5:226-43. [PMID 16041392]
- Okumura K, Mekata E, Shiomi H, Naitoh H, Abe H, Endo Y, et al. Expression level of thymidylate synthase mRNA reflects 5-fluorouracil sensitivity with low dose and long duration in primary colorectal cancer. Cancer Chemother Pharmacol 2008; 61:587-94. [PMID 17520254]
- Zhang X, Diasio RB. Regulation of human dihydropyrimidine dehydrogenase: implications in the pharmacogenetics of 5-FU-based chemotherapy. Pharmacogenomics 2007; 8:257-65. [PMID 17324113]
- Innocenti F, Ratain MJ. Pharmacogenetics of irinotecan: clinical perspectives on the utility of genotyping. Pharmacogenomics 2006; 7:1211-21. [PMID 17184208]
- Kim TW, Innocenti F. Insights, challenges, and future directions in irinogenetics. Ther Drug Monit 2007; 29:265-70. [PMID 17529881]
- Rivory LP, Haaz MC, Canal P, Lokiec F, Armand JP, Robert J. Pharmacokinetic interrelationships of irinotecan (CPT-11) and its three major plasma metabolites in patients enrolled in phase I/II trials. Clin Cancer Res 1997; 3:1261-6. [PMID 9815808]
- Matzow T, Cowen RL, Williams KJ, Telfer BA, Flint PJ, Southgate TD, Saunders MP. Hypoxiatargeted over-expression of carboxylesterase as a means of increasing tumour sensitivity to irinotecan (CPT-11). J Gene Med 2007; 9:244-52. [PMID 17397102]
- Kanzawa F, Saijo N. In vitro interaction between gemcitabine and other anticancer drugs using a novel three-dimensional model. Semin Oncol 1997; 24:S7-8- S7-16. [PMID 9194474]
- Felip E, Rosell R. Testing for excision repair cross-complementing 1 in patients with non-small-cell lung cancer for chemotherapy response. Expert Rev Mol Diagn 2007; 7:261-8. [PMID 17489733]
- Dabholkar M, Vionnet J, Bostick-Bruton F, Yu JJ, Reed E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J Clin Invest 1994; 94:703-8. [PMID 8040325]
- Lord RV, Brabender J, Gandara D, Alberola V, Camps C, Domine M, et al. Low ERCC1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin Cancer Res 2002; 8:2286-91. [PMID 12114432]
- Zhou W, Gurubhagavatula S, Liu G, Park S, Neuberg DS, Wain JC, et al. Excision repair crosscomplementation group 1 polymorphism predicts overall survival in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin Cancer Res 2004; 10:4939-43. [PMID 15297394]
- Bellmunt J, Paz-Ares L, Cuello M, Cecere FL, Albiol S, Guillem V, et al. Gene expression of ERCC1 as a novel prognostic marker in advanced bladder cancer patients receiving cisplatin-based chemotherapy. Ann Oncol 2007; 18:522-8. [PMID 17229776]
- Quinn JE, Kennedy RD, Mullan PB, Gilmore PM, Carty M, Johnston PG, Harkin DP. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res 2003; 63:6221-28. [PMID 14559807]
- Bergman AM, Ruiz van Haperen VW, Veerman G, Kuiper CM, Peters GJ. Synergistic interaction between cisplatin and gemcitabine in vitro. Clin Cancer Res 1996; 2:521-30. [PMID 9816199]
- van Moorsel CJ, Pinedo HM, Veerman G, Bergman AM, Kuiper CM, Vermorken JB, et al. Mechanisms of synergism between cisplatin and gemcitabine in ovarian and non-small-cell lung cancer cell lines. Br J Cancer 1999; 80:981-90. [PMID 10362105]
- Kornmann M, Fakler H, Butzer U, Beger HG, Link KH. Oxaliplatin exerts potent in vitro cytotoxicity in colorectal and pancreatic cancer cell lines and liver metastases. Anticancer Res 2000; 20:3259-64. [PMID 11062751]
- Louvet C, André T, Lledo G, Hammel P, Bleiberg H, Bouleuc C, et al. Gemcitabine combined with oxaliplatin in advanced pancreatic adenocarcinoma: final results of a GERCOR multicenter phase II study. J Clin Oncol 2002; 20:1512-8. [PMID 11896099]
- Faivre S, Raymond E, Woynarowski JM, Cvitkovic E. Supraadditive effect of 2',2'- difluorodeoxycytidine (gemcitabine) in combination with oxaliplatin in human cancer cell lines. Cancer Chemother Pharmacol 1999; 44:117-23. [PMID 10412945]
- Moore MJ, da Cunha Santos G, Kamel-Reid S, Chin K, Tu D, Parulekar W, et al. The relationship of K-ras mutations and EGFR gene copy number to outcome in patients treated with Erlotinib on National Cancer Institute of Canada Clinical Trials Group trial study PA.3. J Clin Oncol, 2007 ASCO Annual Meeting Proceedings Part I. Vol 25, No. 18S (June 20 Supplement), 2007:4521.
- Kindler HL, Niedzwiecki D, Hollis D, Oraefo E, Schrag D, Hurwitz H, et al. A double-blind, placebocontrolled, randomized phase III trial of gemcitabine (G) plus bevacizumab (B) versus gemcitabine plus placebo (P) in patients (pts) with advanced pancreatic cancer (PC): A preliminary analysis of Cancer and Leukemia Group B (CALGB). J Clin Oncol; 2007 ASCO Annual Meeting Proceedings Part I. Vol 25, No. 18S (June 20 Supplement), 2007:4508.
- Lee J, Jang KT, Ki CS, Lim T, Park YS, Lim HY, et al. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer 2007; 109:1561-9. [PMID 17354229]
- Yamanaka Y, Friess H, Kobrin MS, Buchler M, Beger HG, Korc M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res 1993; 13:565-9. [PMID 8317885]
- Dong M, Nio Y, Guo KJ, Tamura K, Tian YL, Dong YT. Epidermal growth factor and its receptor as prognostic indicators in Chinese patients with pancreatic cancer. Anticancer Res 1998; 18:4613-9. [PMID 9891528]
- Tzeng CW, Frolov A, Frolova N, Jhala NC, Howard JH, Buchsbaum DJ, et al. Epidermal growth factor receptor (EGFR) is highly conserved in pancreatic cancer. Surgery 2007; 141:464-9. [PMID 17383523]
- Sung FL, Poon TC, Hui EP, Ma BB, Liong E, To KF, et al. Antitumor effect and enhancement of cytotoxic drug activity by cetuximab in nasopharyngeal carcinoma cells. In Vivo 2005; 19:237-45. [PMID 15796181]
- Rocha-Lima CM, Soares HP, Raez LE, Singal R. EGFR Targeting of Solid Tumors. Cancer Control 2007; 14:295-304. [PMID 17615536]
- Mellinghoff IK, Tran C, Sawyers CL. Growth inhibitory effects of the dual ErbB1/ErbB2 tyrosine kinase inhibitor PKI-166 on human prostate cancer xenografts. Cancer Res 2002; 62:5254-9. [PMID 12234993]
- Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 2006; 355:2733-43. [PMID 17192538]
- Safran H, Iannitti D, Ramanathan R, Schwartz JD, Steinhoff M, Nauman C, et al. Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER-2/neu. Cancer Invest 2004; 22:706- 12. [PMID 15581051]
- Zalatnai A, Molnár J. Review. Molecular background of chemoresistance in pancreatic cancer. In Vivo 2007; 21:339-47. [PMID 17436586]
- Talar-Wojnarowska R, Gasiorowska A, Smolarz B, Romanowicz-Makowska H, Strzelczyk J, Janiak A, et al. Clinical significance of K-ras and c-erbB-2 mutations in pancreatic adenocarcinoma and chronic pancreatitis. Int J Gastrointest Cancer 2005; 35:33-41. [PMID 15722572]
- Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol 2004; 22:1430-8. [PMID 15084616]
- Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M, Buckels JA. A double-blind placebocontrolled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer 2002; 87:161-7. [PMID 12107836]

