Keywords
Proximate, Mineral, Composition, Medicinal, Analysis.
Introduction
Salacia senegalensis belong to the
Kingdom-plantae, Order-celastrales, Family
-Celastacea, Genus-Salacia, Speciessenegalensis [1-3]. Salacia senegalensis Lam
(DC) (Figure 1) is an erect or climbing
shrub with white or pale greenish cream petals and orange or yellow flowers. It is
found in tropical forests. It belongs to the
family Celastraceae. The Igbo name is
“Nriaturu” or “Nriatulu”. Traditionally, the
extract of its leaf is used in malaria
treatment, as a lotion for sick children and in
the treatment of skin problems like eczema
by the people of South-East zone of
Nigeria [4]. A decoction or alcohol extract of
Salacia senegalensis leaf is used by the Orji
people of Owerri North local government
area (L.G.A), Imo State, South-East Nigeria
as a remedy for malaria [4]. The antimalarial
activities of its leaf have been reported [5].
Also [6] reported the terpenes composition of
its leaf. However, scientific data on the
proximate, vitamins and mineral
composition of its leaf is yet to be reported,
therefore, this work was aimed at analyzing
proximate, vitamins and mineral
composition of Salacia senegalensis leaves.
Figure 1. Salacia senegalensis
Figure 2. Chromatogram of the vitamins composition of Salacia senegalensis leaves.
Materials and Methods
Determination of the moisture and dry
matter content
The AOAC Official Method 967.03 [7]
was adopted.
Principle
When tissues are subjected to
prolonged exposure to high temperature of
about 100-1050C, they gradually lose water
and consequently, lose weight. This weight
loss continues until they attain a constant
weight when they can no longer lose water.
This constant weight is the dry matter
content while the weight loss is the moisture
content.
Materials
These include (i) the sample. (ii)
Crucible, dessicator, weighing balance
(Mettler Toledo AB204; Mettler Toledo,
Switzerland) and oven (Plus II Sanyo,
Gallenkamp PLC, England).
Procedure
A fire-polished empty crucible was
allowed to cool in the dessicator, after which
its weight was taken. Then 2g of the sample
was placed in the crucible. The crucible and
content were then placed in the oven at
105oC, for 3 hr, and was weighed regularly
until a constant weight was obtained after
cooling. This procedure was repeated for
triplicate samples.
Calculation
Moisture content (%) = weight of
fresh sample (g) – weight of dry sample (g)
X 100 / weight of fresh sample (g).
Dry matter content (%) = weight of
dry sample (g) X 100 / weight of fresh
sample (g).
Determination of the ash content
The AOAC Official Method 942.05 [7]
was adopted.
Principle
When heated to high temperature of
between 560 and 600°C, the organic
portions of samples are converted to volatile
compounds (e.g. oxides, etc.), leaving a
grayish-white residue of the inorganic
portion. Ash is determined by weighing the
resulting inorganic residue.
Materials
These include (i) the sample. (ii)
Crucible, dessicator, weighing balance
(Mettler Toledo AB204; Mettler Toledo,
Switzerland), timer and muffle furnace.
Procedure
Two grammes of the sample was
added to a pre-weighed, clean, fire-polished
empty crucible, and placed in the muffle
furnace at 600°C. After 7 hr, when only a
grayish-white residue of the sample was left,
the crucible and content was removed and
cooled in a dessicator. The weight of the crucible and residue was taken. This
procedure was repeated for triplicate
samples.
Calculation
Ash content (%) = weight of residue
(g) X 100 / weight of fresh sample (g).
Determination of the total carbohydrate
content
The Anthrone Method as reported
by [8] was adopted.
Principle
Perchloric acid hydrolyzes the
available carbohydrates to sugar (glucose)
which is then dehydrated by concentrated
H2SO4, to furfural or furfural derivatives
that react with anthrone to give a blue-green
complex. The blue-green solution formed,
has absorption maxima at 630nm.
Materials
These include (i) the sample. (ii)
52% (v/v) perchloric acid in concentrated
H2SO4; 0.1% standard glucose; 0.2%
Anthrone Reagent in concentrated H2SO4.
(iii) Spectrophotometer [Model 752S
(Spectrumlab)], weighing balance (Mettler
AB204; Mettler Toledo, Switzerland), water
bath, timer, volumetric flask, glass filter,
pipette, cuvette, test tubes and test tube rack.
Procedure
The procedure involved
deproteination, extraction and hydrolysis of
the sample. To 0.1 g of the ground sample in
a 25ml volumetric flask was added 1.0ml of
distilled water and 1.3ml of 52% perchloric
acid a lask was
stoppered and the mixture allowed standing
for 20 min, with shaking to ensure complete
hydrolysis. The content was then made up to
the 25ml mark with distilled water and
allowed to stand for 30 min to enable
extraction, after which it was filtered through a glass filter. The filtrate (0.1ml)
was used as test solution. Three test tubes
were set up labeled T1 (blank), T2 (glucose
standard) and T3 (test sample). T1 contained
0.1ml of distilled water; T2 contained 0.1ml
of standard glucose solution while T3 contained 0.1ml of test filtrate. To each tube
was added 0.9ml of 0.2% Anthrone Reagent.
The contents were thoroughly mixed, placed
in boiling water bath for 12 min, cooled and
absorbance read at 630 nm in the
spectrophotometer. This procedure was
repeated for triplicate samples.
Calculation
% Carbohydrate content =
absorbance of filtrate X C X 25 / absorbance
of standard.
Where c = concentration of
standard (mg/ml); 25 = final volume of
hydrolyzed sample (ml).
Determination of the protein content
The determination of the protein
content was based on the semi-macro
Kjeldahl method as described by [9].
Principle
In this method, the product was
digested with concentrated sulphuric acid
using copper sulphate as a catalyst to
convert organic nitrogen to ammonium ions.
Alkali was added and the liberated ammonia
distilled into an excess of boric acid
solution. The distillate was then titrated with
hydrochloric acid to determine the ammonia
absorbed in the boric acid.
Materials
These include (i) the sample. (ii)
Kjeldahl catalyst [Na2SO4 and
CuSO4.5H2O, (10:1 w/w)]; concentrated
H2SO4; 40% NaOH; 2% boric acid; 0.1 M
hydrochloric acid; double indicator [2 parts
of 0.2% (w/v) methyl red in ethanol to 1 part
of 0.2% (w/v) methylene blue in ethanol].
(iii) Weighing balance, Kjeldahl digestion
flask, 100ml volumetric flask, beaker,
burette, pipette or measuring cylinder,
Buchner funnel and anti-bumping chips.
Procedure
Digestion
To 0.1 g of the sample in a 100 ml
Kjeldahl digestion flask, was added 3 g of
Kjeldahl digestion catalyst, 25 ml of
concentrated sulphuric acid and a few antibumping
chips, and mixed by greatly
swirling the mixture. The flask was fitted to
reflux condenser and gently heated until
foaming had ceased, and the contents
became completely liquefied. Then, the
content of the flask as boiled vigorously,
with occasional rotation of the flask, until
the colour of the digest change from ash to
blue-green or pale green colour. The flask
was allowed to cool and its contents were
quantitively transferred into a 100ml
volumetric flask, where it was dilute to the
100ml mark with distilled water.
Distillation
Twenty milliliters of this diluted
digest was transferred into a 150ml
distillation flask. The flask into which a few
anti-bumping chips have been added was
connected to a condenser whose receiver
was attached to a Buchner funnel immersed
in a 400 ml beaker containing 10ml of 2%
boric acid solution masked with 2 drops of
double (methyl red-methylene blue)
indicator. Then, 25ml of 40% NaOH was
added to the flask using a syringe.
Distillation was stopped when the volume in
the beaker was about the same as the
original volume, and the colour of the boric
acid in the receiving flask changed from
purple to pale green. The ammonia was
liberated into the boric acid solution. The
distillation unit was dismantled and rinsed
with distilled water.
Titration
The distillate (boric acid-ammonia
solution) was titrated with 0.1 M
hydrochloric acid, until the colour changed
to pink, which marked the end of titration.
Thetitre was recorded and this was used to
determine the nitrogen content from which
the protein value was calculated by
multiplying with the nitrogen conversion
factor, 6.25.
Calculation
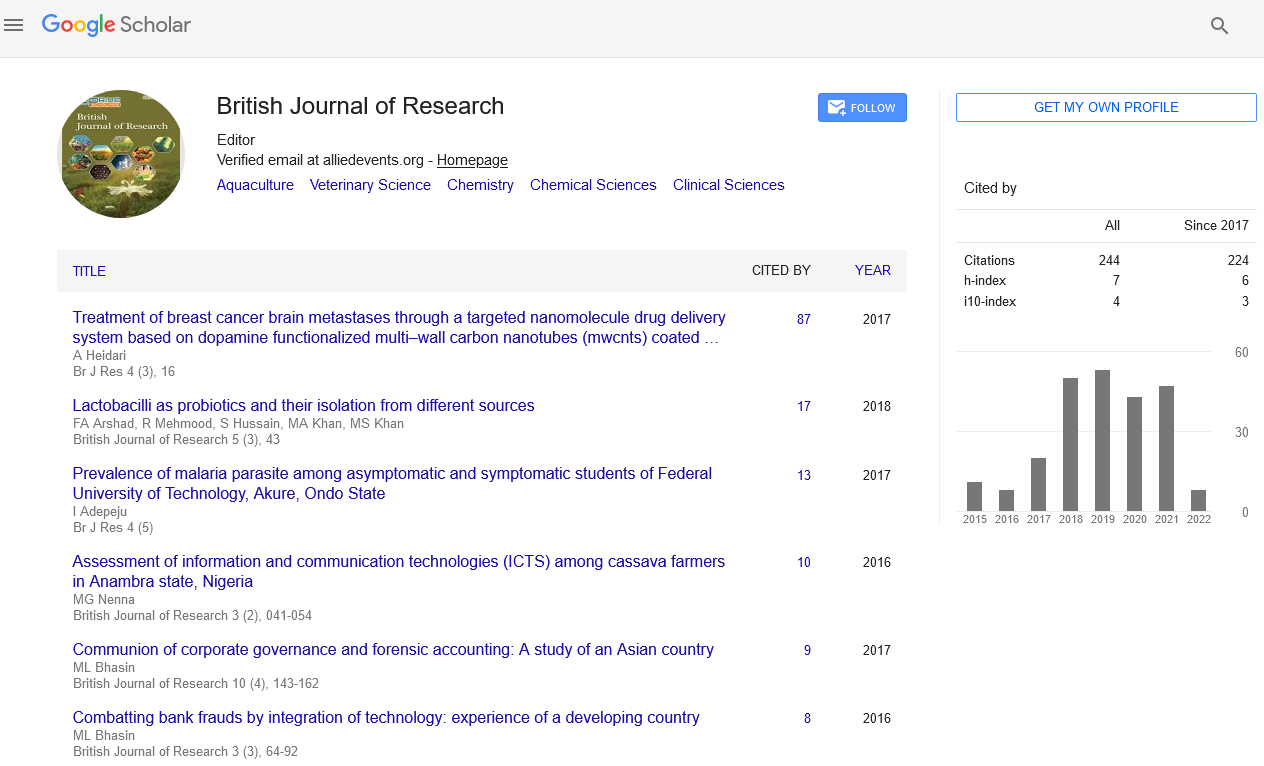
Kjeldhal nitrogen (%) = 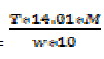
Where T= titre value (ml); w =
sample weight (g); M =molar concentration
(moles/L) of the acid; 14.01 = atomic weight
(g) of N; and 10 = factor to convert mg/g to
percent.
Crude protein content (%) = %
Kjeldhal nitrogen * 6.25.
Where 6.25 = factor to convert N to
protein.
Determination of the lipid content
The AOAC Official Method 920.397
was adopted.
Principle
The lipid content of the sample was
extracted with anhydrous diethyl ether and
weighed.
Materials
These include (i) sample. (ii)
Anhydrous diethyl ether. (iii) Soxhlet
extractor, round bottomed flask, oven,
Whatman No. 1 filter paper, weighing
balance, dessicator, timer and heating
mantle.
Procedure
Three grammes of the ground were
transferred into a thimble and dried for 3 hr
at 100 °C. The extraction thimble containing
the dried samples was in turn inserted into
the extraction chamber. The extractor and its contents were fixed to a pre-weighed empty
round bottomed flask which was in turn
placed over an electro-thermal heater. Then,
300 ml of anhydrous diethyl either was
poured into the flask through the extractor
and the condenser was fixed. The sample
was then extracted for 6 hr, after which the
solvent was distilled off, leaving the lipid in
the flask to cool in a dessicator. The flask
and content were then weighed.
Calculation
:. Crude lipid content (%) = 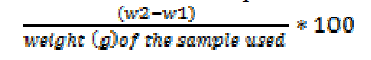
Where w1 = weight (g) of empty
flask; w2 = weight (g) of flask and lipid.
Determination of the fiber content
This was based on AOAC Official
Method 973.18 [7].
Principle
A fat-free sample was treated with
boiling sulphuric acid and subsequently with
boiling NaOH. The residue after subtraction
of the ash is regarded as fiber.
Materials
These include (i) the sample. (ii)
1.25% H2SO4 solution; 1.25% NaOH
solution; 1% HCI solution; petroleum ether
(60-80 oC); absolute ethanol (iii) Oven,
weighing balance, water bath, muffle
furnace, dessicator, timer, crucible, spatula,
Whatman No. 1 filter paper, conical flask,
Buchner funnel, soxhlet extractor and reflux
condenser.
Procedure
Two grammes of the ground sample
(in triplicate) were transferred into a crucible
and dried for 3 hr at 100 °C. The dried
samples were defatted by extraction with 10
ml of petroleum ether (60-80 °C) and air
dried. The defatted sample was transferred
into a 1000 ml conical flask into which 20 ml of 1.25% H2SO4 was added and mixed
properly. An aliquot (190 ml) of boiling
H2SO4 was added into the flask and mixed
properly such that a cream was produced.
This flask was fitted to reflux condenser and
heated before its content was rapidly poured
into a shallow layer of hot water contained
in a hot Buchner funnel prepared with a wet
12.5cm filter paper. Filtration was done by
suction and the rate of suction adjusted such
that filtration was completed within 10 min.
The residue was washed free of acid with
hot distilled water and quantitatively
transferred into 100 ml volumetric flask
using 200 ml of boiling 1.25% NaOH
solution. Refluxing was done for 30 min and
the filtrate was allowed to cool for a minute
and filtered under suction. The residue was
washed with several portions of boiling
water (distilled) followed by 1% HCI,
finally with boiling water until the wash was
acid free. Further washing was done twice
with ethanol and twice with petroleum ether.
The residue was quantitatively and carefully
transferred (using a clean spatula) into a
weighed crucible, which was dried in an
oven at 105 °C for one hour. Then, it was
cooled in a dessicator and weighed. The dry
residue was then placed in a furnace at 630
°C for 3 hr. the crude fiber content was
calculated as well as the percentage crude
fiber. The weight of the ash was subtracted
from that of the residue to obtain the weight
of the fiber. This procedure was repeated for
triplicate samples.
Calculation
Fiber content (%) = 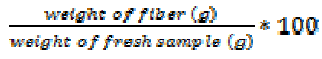
Determination of the energy content
The calorific value was obtained by
multiplying the values of crude protein,
crude fat and total carbohydrates by the
Atwater factors 4, 9 and 4, respectively,
taking the sum of the products and expressing the result in kilocalories per 100
g sample as reportedby [10-14].
Determination of vitamin composition
The samples were extracted
following the method of [15].
Principle
Esterified extracts are more readily
separated by gas chromatography. The
effluent can be identified by pulse flame
photometric detector.
Materials
These include (i) the sample. (ii)
Absolute ethanol; redistilled methanol;
chloroform; anhydrous sodium sulphate;
standards; Boron Trifluoride Reagent (7 %
BF3 (w/w) in methanol, made from
commercially available 14 % BF3 solution).
(iii) Soxhlet extractor, Janke and Kunkel
(IKA-LABORTECHNIC) grinder, timer,
weighing balance, rotary evaporator, oven,
beakers, and GC system with pulse flame
photometric detector.
Procedure
The dirt free sample was weighed
and pulverized into fine powder, using Janke
and Kunkel (IKA-LABORTECHNIC)
grinder. One gramme of the pulverized
sample was homogenized in 1 mL, of
ethanol, and extracted by refluxing with 10
mL of re-distilled methanol, for 6 hr at very
low temperature. The process was repeated
twice, using fresh solvents, ensure that most
of the water soluble vitamins in the
pulverized sample was homogenized in 1
mL of ethanol, and extracted by refluxing
with 10 mL of chloroform, for 6 hr at very
low temperature. The process was repeated
twice, using fresh solvents, to ensure that
most of the fat soluble vitamins in the
pulverized sample were removed. The
extract was then evaporated to dryness on a
rotary evaporator. To residue was added 4.00 ml of 7 % BF3 (Boron Trifluoride)
Reagent, and heating for 45 min in an oven
at 100 oC. It was cooled to room
temperature, and 1.0 g of anhydrous Na2SO4
was added to remove water, after which is
was subjected to gas chromatography
analysis, using pulse flame photometric
detector, for the determination of the
component vitamins.
Standard solutions were prepared
and the linearity of the dependence of
response on concentration was verified by
regression analysis. Identification was based
on comparison of retention times and
spectral data with standards. Quantification
was performed by establishing calibration
curves for each compound determined, using
the standards.
Chromatographic Conditions
Chromatographic analysis was
carried out using a GC Model HP6890
powered with HP Chemstation Rev A 09.01
(1206) software (to identify and quantify
compounds), with pulse flame photometric
detector. The capillary column was a ZB-5
Column (30 m x 0.32 mm x 0.25μm film
thickness). The oven temperature was held
at 30 oC for 2 min, and then programmed at
4 oC/min for 15 min before changing to 15
oC/min for 2 min. Hydrogen was used as a
carrier gas. The hydrogen column, hydrogen
and compressed air pressures were
respectively 25 psi, 22 psi and 28 psi. The
injector and detector temperature were kept
at 250 and 300 oC, respectively; split ratio,
2:1, 0.2μL of the sample was injected.
Determination of mineral composition
Atomic Absorption Spectrophotometry
was used in mineral determination.
In accordance with the
manufacturers’ instructions, the instrument,
Perkin Elmer atomic absorption
spectrophotometer (AAS) PE 600c, was set
up. It was switched on and allowed for 15minutes equilibration period. It was then
set up by putting in place the hollow cathode
tube appropriate for the element being
determined. The monochromator was set at
the wavelength of the element being
determined, Example, 325nm for Copper.
With these in place, the instrument was
flushed by aspirating distilled de-ionized
water in it. It was then calibrated at zero
with the reagent blank (HCl solution).
Meanwhile, standard solutions of each of the
test elements being determined were
prepared separately and diluted in series
according to chosen concentrations. The
diluted solutions of the test elements were
aspirated in-turns into the instrument and
their respective absorbencies were recorded
and plotted into a standard curve. Then the
sample extracts were aspirated in-turns into
the instrument and their absorbance were
also recorded. The standard curve was used
to extrapolate contents of the test element in
the sample extract.
The general formula below was used
to quantify the content of each element in
the sample,
A (mg/100 g) = 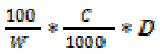
Where: A = element being determined,
W = weight of sample used,
C = concentration (in ppm) obtained from
the standard curve,
D = Dilution factor applied
Results
The proximate composition (%) of the
leaf of Salacia senegalensis (S. s.) is shown in Table 1. The results showed that carbohydrate
was highest (57.28 ± 0.01 %) followed by
crude fibre(24.85 ± 0.15 %), moisture (22.27
± 0.04 %), protein (18.00 ± 0.10 %), lipids
(1.82 ± 0.01 %) and the least was ash (0.63 ±
0.02 %) as presented in Table 1. The energy
value was 317.5kcal/100g as shown in Table 1.
Table 1. Proximate composition of Salacia senegalensis leaves
The concentrations of the vitamins
assayed as are shown in Table 2. The results
showed that vitamin C in the leaf was the
highest (45.01 mg/100g), followed by vitamin
B3(0.14 mg/100g), vitamin B2 (0.08 mg/100g)
and vitamin B1(0.03 mg/100g), while, very
low levels of vitamins E (0.008 mg/100g),
B9(folate) (0.007 mg/100g) and B6 (0.007
mg/100g) respectively were detected.
Table 2. Vitamins composition of Salacia senegalensis leaves
The concentrations (mg/100g) of
minerals present in the leaf of Salacia
senegalensis are shown in Table 3. Calcium
had the highest value (27.31 mg/100g),
followed by magnesium (16.01 mg/100g),
sodium (11.83 mg/100g), iron (11.75
mg/100g), potassium (9.57 mg/100g),
manganese (3.05 mg/100g) and zinc (1.01
mg/100g). Then, copper (0.95 mg/100g),
followed by nickel (0.02 mg/100g), and
chromium (0.01 mg/100g).While lead and
cadmium, were not detected (n.d).
Table 3. Mineral composition of Salacia senegalensis leaves
Discussion
The results of proximate composition
of Salacia senegalensis leaves are shown in Table 1. The moisture content of Salacia
senegalensis leaves was 22.27 ± 0.04 %. The
moisture content of any food is an index of its
water activity [16,17], and is used as a measure of
stability and susceptibility to microbial
contamination [18]. Its moisture content is within
the range of required value as safe storage
limit for plant food materials [19]. This indicates
that the leaves can be stored for a long time
without the development of mould.
The crude protein of the Salacia
senegalensis leaves was 18.00 ± 0.10 % per
100g which is comparable to the daily protein
requirement of 23-56 g [10,14].
Ash content is a measure of the total
mineral content of a food [20]. The leaves had a
low value of 0.63 ± 0.02 % ash (mineral)
content. Mineral is required by the body for
proper physiological functioning.
The carbohydrate content was high
(57.28 ± 0.01 %) which is comparable to that of Vitex doniana leaves (67.00%) [21]. This
means it could serves as a sources of energy.
The crude fibre content was 24.85 ±
0.15 % which is higher than 15.00 % reported
for V. doniana leaves [21] or 19.80% for
Asparagus officinalis tem [22]. Epidemiological
evidence suggest that increased fibre
consumption may contribute to a reduction in
the incidence of certain diseases including
colon cancer, coronary heart diseases,
diabetes , high blood pressure, obesity, and
various digestive disorders [23,24].
Dietary fibre has been associated with
alterations of the colonic environment that
protect against colorectal diseases. Fibre may
also provide protection by increasing faecal
bulk, which dilutes the increased colonic bile
acid concentrations that occurs with a high fat
diet [23,25].
The fibre recommended dietary
allowance (RDA) values for children, adults,
pregnant and breast feeding mothers are 19-
25%, 21-28%, 28% and 29% respectively [20].
The crude fat of 1.82 ± 0.01 % was
obtained on analysis of Salacia senegalensis
leaves. This is lower than that reported (3%)
for V. doniana by [26].
Fats/oils are very important in human
health as it serves as sources of energy and
components of biological membranes [27].
Energy value of the Salacia
senegalensis leaves was 317.5kcal/100g. This
again pointed the caloric value of the leaf.
Salacia senegalensis leaves had high
vitamin C content (45.01 mg/100g), which is
about 99.37% of the total vitamin
composition of the leaves. Vitamin C
(ascorbic acid) is generally involved in
protein metabolism and collagen synthesis [20].
Though this value is lower than the RDA
value (60mg/100g) for adult [28], it could
supplement for the daily needs. Ascorbic acid
is a potent anti-oxidant that serves to
regenerate vitamin E from it’s oxidized
byproduct [29].
Vitamin B1 (thiamin), B2 (riboflavin)
and B6 (pyridoxial) are associated in
macronutrient metabolism and are present in
Salacia senegalensis leaves at a low level of
0.03mg/100g, 0.08 mg/100g and 0.01
mg/100g respectively as shown in Table 3.
These values are relatively low compared
with RDA values of 1.2, 1.4, and 1.5mg/100g
respectively20.
The vitamin E content of Salacia
senegalensis leaves was 0.01mg/100g
(0.02%). Vitamin E serves as an antioxidant
of polyunsaturated fatly acid in cell
membranes and sub-cellular structures [30]. It
influences cellular response to oxidative stress
through signal transduction pathways [31].
The vitamin B3 (niacin) content was
0.14mg/100g (0.32%). This value is relatively
lower than the RDA of 14mg/100g [32].
Vitamin B3 is a precursor to nicotinamide
adenine nucleotide and nicotinamide adenine
dinucleotide phosphate, which serves as
electron and proton acceptors, respectively [32].
The leaves of Salacia senegalensis
had 0.01 mg/100g of vitamin B9 (folate),
about 0.02% of the total vitamin as shown in Table 3. Folate is required for synthesis of
purines and pyrimidines that are needed for
DNA production and erythropoiesis32. This
value is lower than the RDA of 0.4 mg [32].
Table 3 showed that the Salacia
senegalensis leaves had high calcium content
(27.3 mg/100g). Calcium forms component of
bones and teeth, necessary blood clothing and
muscle contraction [20]. This value is lower than
71.0mg/100g reported in the leaves of V.
doniana [21].
Salacia senegalensis leaves had 16.01
mg/100g of magnesium. Magnesium is an
important element in connection with
circulatory diseases and calcium metabolism
in bone [33]. The value (16.01 mg/100g)
reported here is lower than 124mg/100g
reported by [34] and 45.0mg/100g for V. doniana
leaves [21].
The Salacia senegalensis leaves had
11.83mg/100g sodium and 9.57mg/100g
potassium. Sodium content in combination
with potassium is involved in maintaining
proper acid-base balance and in nerve impulse
transmission in the body [35]. The variation of
potassium to sodium content in this work is of
significant importance particularly to a
hypertension patient [19]. These value obtained
here are lower than the ones obtained from
the leaves of V. doniana [26] and [21].
The iron content of Salacia
senegalensis leaves was 11.75mg/100g. Iron
is essential micronutrient for haemoglobin
formation, normal functioning of central
nervous system (CNS) and in the oxidation of
carbohydrate, protein and fat [36]. Since it had
significant amount of iron, its consumption
may be encouragement particularly for
menstruating and lactating women [20].
The leaves of Salacia senegalensis
had 3.05mg/100g of manganese. Manganese
is a cofactor of hydrolase, decarboxylase, and
transferase enzymes37. This value is higher
than that of cashew nut [38] and [39].
The zinc content of Salacia
senegalensis leaves was 1.01 mg/100g. Zinc
function as cofactor of many enzymes like
lactate dehydrogenase, alcohol
dehydrogenase, glutamic dehydrogenase,
alkaline phosphatase, carbonic anhydrase,
carboxypeptidase, superoxide dismutase,
retinenereductase, DNA and RNA
polymerase [40]. This value is lower than that of
cashew nuts [38,39].
The leaves of Salacia senegalensis
had 0.93mg/100g of copper, which is grossly
lower than 65.0mg/100g reported by [19] of V.
doniana leaves. This value is lower than the
RDA (1.5-3.0) mg/day) of copper [20]. Copper is
a constituent of enzymes (cofactor) like
cytochrome c oxidase, amino oxidase,
catalase, peroxidase, ascorbic acid oxidase,
plasma monoamine oxidase, erythrocuprin
(ceruloplasmin), lactase, uricase, tyrosinase,
cytosolic superoxide dismutase and it plays a role in iron absorption [41]. It is essential for
haematologic and neurologic systems [42].
Salacia senegalensis leaves had
grossly low levels of chromium and nickel
respectively as shown in Table 3. This shows
that Salacia senegalensis leaves could not be
a good source of chromium and nickel
respectively. Lead and cadmium was not
detected.
Conclusion
The result indicates that Salacia
senegalensis leaf could supplement for
carbohydrate, crude fibre and protein, while
its relative high Ca and Mg values could be
used for the management of bone ailments.
Also it could serve as source of antioxidant
due to its high content of vitamin C.
References
- Loesener, T (1942). Celastraceae, In: Engler A, Harms H, Mattfeld J, Die. Naturlichen Pflanzenfamilien. Duncker, Humlot, Berlin. 20b: 87–197.
- Simmons, M.P (2004). The Families and Genera of Flowering Plants. Celastraceae, in: Kubitzki K edn: Springer, Berlin. 29–64.
- Chawla, A; Singh, S; Sharma, A. K (2013). Salacia oblonga Wall: A Review on its Pharmacognostic, Phytochemical and Pharmacological Aspects, International Journal of Research in Pharmaceutical and Biomedical Sciences, 4 (4): 1215-1228.
- Nigeria National Medicine Development Agency (NNMDA) (2011).Salacia senegalensis, Medicinal plants of South- East Zone, 1: 67.
- Adumanya, O.C.U; Uwakwe, A.A and Essien, E.B (2014a). Antiplasmodial Activity of Methanol Leaf Extract of Salacia senegalensis Lam (DC) in Albino Mice infected with Chloroquine- Sensitive Plasmodium berghei (NK65). International Journal of Ethnopharmacology. 1(1): 002-006.
- Adumanya, O.C.U; Uwakwe, A.A; Essien, E.B. (2014b). Essential Oil Composition (Terpenes) of Salacia senegalensis Lam (DC) Leaf. British Journal of Research, 1(2):026-034.
- AOAC International (2006). Official Methods of Analysis of the AOAC (W. Horwitiz, Editor). 18thedn. Washington DC, USA: AOAC International.
- Plummer, D.T (1978). An Introduction to Practical Biochemistry. 2ndedn. London: McGraw-Hill.
- Pellet, P.L. and Young, V.R. (Eds.), (1980). Nutritional Evaluation of Protein Foods. Report of Working Group Sponsored by the International Union of Nutritional sciences and the United Nations University World Hunger Programme. The United Nations University: United Nations University Press. WHTR-3/UNUP-129. ISBN: 92- 808-0129-5.
- FAO/WHO/UNU (1991). Energy and Protein Requirements: Report of a Joint FAO/WHO/UNU Expert consultation WHO Technical Report. Series724.ISSN 0512-3054 https://www.fao.org/docrep/ t0207e/T0207Eo8. Httm.
- Onyeike, E.N. and Ehirim, F.C. (2001). Chemical and Sensory Evaluation of Melon fungus (Pleurotustuberregium) and Melon fungus Cake. Journal of Biochemistry & Molecular Biology, 16(1); 77-81.
- Onyeike, E.N. and Acheru, G.N. (2002). Chemical composition of Nigerian oil seeds and physicochemical properties of the oil extracts. Food Chemistry, 77(4):431-437.
- Onyeike, E.N; Ihugba, A.C and George, C. (2003). Influence of Heat Processing on the Nutrient Composition of Vegetable Leaves Consumed in Nigeria. Plant Foods for Human Nutrition, 58(3):1-11.
- Chaney, S.G (2006). Principles of Nutrient 1: Macronutrients. In T.M. Devlin (Ed.), Textbook of Biochemistry, with clinical correlation, 6thedn (pp, 1071-1090). New York John Wiley and sons.
- Zhao, B; Tham, S.Y; Lu, J; Lai, M.H; Lee, L.K.H. and Moochhala, S.M. (2004). Simultaneous Determination of Vitamins C, E and β-carotene in Human Plasma by High-Performance Liquid Chromatography with Photodiode-array Detection. Journal of Pharmacy & Pharmaceutical Science, 7(2): 200-204.
- Olutiola, P.O; Famurewa, O. and Sonntag, H.-G. (1991). An Introduction to General Microbiology, a Practical Approach. Germany: Heidelberger Verlagasanstalt and Druckerei GmbH Heidelberg.
- Pearson, A (1994). Vitamins in fruits. The Biochemistry of fruit and other products. Academic press; New York, 369-384.
- Uraih, N and Izuagbe, Y. (1990). Public Health Food and Industrial Microbiology Nigeria: Uniben Press.URL:http//www.fao.org/docrep/00 5/ac45/eob.htm#fn/ll. URL:https://www. spectracell.com/media/patient-brochureheart- disease.pdf.
- Umar, K.S; Hassan, L.G and Ado, Y (2007). Mineral composition of Detariummicrocarpum Grown in Kwatarkwashi, Zamfara State, Nigeria, Inter, J. Pure Appl. Sci. 1(2): 43-48.
- Vunchi, M.A; Umar, M.A; King, A.A; Liman, G.J and Aigbe, C.O (2011). Proximate, Vitamins and Mineral composition of Vitexdoniana (black plum) fruit pulp. Nigerian Journal of Basic and Applied Science, 19(1):97- 101.
- Nnamni, C.V; Oselebe, H.O and Agbatutu, A (2009). Assessment of Nutritional value of three underutilized indigenous leafy vegetables of Ebonyi State, Nigeria. Afr. J. Biotech, 8(9):2321-2324.
- Ali, A (2009). Proximate and mineral composition of the Marchubeh (Asparagus officinalis) in Iran. J. Diary Food Sci. 4(2):145-149.
- Food and Agriculture Organization (1990). Roots, Tubers, Plantains and Bananas in Human Nutrition. FAO Corporate Document Repository, Rome https://www.fao.org/docrep/t0207e/T020 7Eo8.httm.
- Scientific Advisory Committee on Nutrition. (2008). Draft SCAN position statement on dietary fiber and health and the dietary fiber definition-August 2008. SACN/08/20. https://www.sacn.gov.uk/ pdfs/finai draft sacnstatement on dietary fibre for website.
- Dillard, C.J and German, J.B (2000). Phytochemicals: Nutraceuticals and Human Health. Journal of the Science of Food and Agriculture. 80(12): 1744- 1756.
- Agbede J.O and Ibitoye, A. A. (2007). Chemical composition of Black plum (Vitexdoniana); an underutilized fruit. J. Food Agric. Env. 5(2): 95-96.
- Michelle, A; Hopkins, P.J.J; McLaughlin, C.W; Johnson, S.E; Warner, M.Q; Lahart, D.F and Wright, J.D (1993). Lipids, Human Biology, and Health, Eagle Wood Cliffs, New Jersey, USA-Prentice Hall, p1176-1180.
- Rod, R.S; Trend, S and Philip, T.D.A (1996). Essentials of Anatomy and Physiology 2nd edition, McGraw Hell Companies, 467-469.
- Sen, C, K and Hanninen, O. (1994).Physiological Antioxidants. In: Sen, CK Packer L, Hanninen O, eds Exercise and Oxygen Toxicity. Amsterdam: Elsevier, 89.
- Machlin, I.J and Bendich, A (1987). Free Radical Tissue Damage: Protective Role of Antioxidant Nutrients. FASEB J. 1:441.
- Azzi, A; Boscobonik, D and Hensey, C. (1992). The Protein Kinase C family. Eur. J. Biochem. 208:547.
- Lukaski, H.C (2004). Vitamin and Mineral Status: Effects on Physical Performance. Nutrition, 20: 632-644.
- Ishida, H; Suzuno, H; Sugiyaman, N; Innami, S and Maekawa, A (2000). Nutritional Evaluation of chemical component of leaves stalks and stem of sweet potatoes (Ipomeabatataspon). Food Chem., 68:359-367.
- Robert, H.G; Dorothy, J.V; Cassius, L; Louis, E.G; Garret, C.S; Andrezej, P and Mark, M (1997). Amino acid, fatty acid and mineral composition of 24 indigenous plants of Burkino Faso. J. Food Comp. Anal. 10:205-217.
- Adeyeye, E.I (2002). Determination of the chemical composition of the nutritionally valuable parts male and female common West African fresh water crab. SudnanautesafricanusInt. J. Food Sci. Nutr., 53:189-196.
- Adeyeye, E.I and Otoketi, M.K.O (1999). Proximate composition and some nutritionally valuable minerals of two varieties of Capsicum annum (Bell and Cherry peppers). Discovery and Innov., 11(1&2): 75-81.
- Murray, R.K; Granner, D.K; Mayes, P.A and Rodwell, V.W (2000). Harper’s Biochemistry, 25th Edition, McGrawHill, Health Profession Division, USA.
- Nandi, B.K (1998). Cashew nut nutritional aspects. In M.K. Papademetroiu and E.M. Herath (Eds.). Integrated production practices of cashew in Asia. Food and Agriculture Organization of the United Nations Regional office for Asia and the Pacific Bangkok, Thailand (FAO/RAP Publication: 1998/12).
- Nutrition Data, (2009). Know what you eat: Nuts, cashew nuts, oil roasted, without salt added. Retrieved March 18, 2011 from https://nutritiondata.self.com/ facts/nut-and-seedproducts/3094/2.
- Soetan, K.O; Olaiya, C.O and Oyewole, O.E (2010). The importance of mineral elements for humans, domestic animals and plants; A review. African Journal of Food Science 4(5):200-222.
- Chandra, R.K (1990). Micro-nutrients and immune functions: An overview Annal New York Acad. Sci., 587:9-16.
- Tan, J.C; Burns, D.L and Jones, H.R (2006). Servere ataxia, myelopathy and peripheral neuropathy due to acquired copper deficiency in a patient with history of gastrectomy. J. Paenteral Mutr. 30:446-450.