Keywords
Neoplasms; Pancreas; Pulmonary Blastoma
INTRODUCTION
Pancreatoblastoma is a rare tumor of the pancreas and usually occurs in pediatric patients. Since it was first described in 1957, close to 200 cases have been described and most were in the pediatric population [1]. In 1986, Palosaari et al. described the first case of pancreatoblastoma in an adult [2]. Only 15 case reports of pancreatoblastoma in adults have since been published. Most of these cases describe patients with a short-term survival of just several months after diagnosis. The cases that do report patients with disease-free survival had followed the patients for at most three years at the time the articles were published. Therefore, the longterm prognosis and best treatment strategy for these patients remains uncertain. We present a case of an adult patient with pancreatoblastoma who remains disease free five years after his first resection, despite the presence of liver metastasis and positive microscopic tumor margins after surgery. To our knowledge, this case represents the longest reported period of disease-free survival for an adult patient with metastatic pancreatoblastoma and supports an aggressive surgical and chemotherapeutic approach to treatment.
CASE REPORT
The patient is a 33-year-old man who noted right upper quadrant abdominal pain and a 9 kg weight loss over the preceding two months. Past medical history included postpubertal gynecomastia with breast tissue resection as a young adult and peptic ulcer disease treated with oral antacids. Physical exam revealed a mildly tender right upper quadrant abdominal mass. Computed tomography (CT) of the abdomen demonstrated a large mass in the right lobe of the liver. No pancreatic lesions were observed. A CT-guided fine needle aspiration biopsy suggested a diagnosis of neuroendocrine carcinoma. He had no laboratory abnormalities, including normal liver function tests, CA 19-9, and CEA levels. At exploration, a firm 12 cm mass in the right lobe of the liver was palpated. Intraoperative ultrasound of the liver revealed no other lesions and no lymphadenopathy was appreciated. However, in the pancreas, two distinct masses were felt. One mass was located in the body of the pancreas and the other was a hypoechoic mass in the head near the portal vein. Multiple core-needle biopsies of the pancreas were performed under ultrasound guidance. The frozen section showed only evidence of chronic pancreatitis and the patient underwent a standard right hepatic lobectomy (segments 5-8) and did well post-operatively.
Grossly, the resected liver mass, measuring 12x11x7 cm, was well circumscribed and solid with areas of necrosis and hemorrhage. Microscopically, the tumor was highly cellular and composed of variably-sized nodules or lobules of tumor cells separated by fibrous tissue. The tumor cells were predominantly arranged in sheets of rosettelike structures with lumens resembling the pancreatic acini (Figure 1). Less commonly, there were tubular, trabecular, or solid patterns. The tumor cells were uniform and had a moderate amount of fine eosinophilic granular cytoplasm. The nuclei were uniformly round or oval with stippled chromatin and small nucleoli. Mitotic activity was easily identified with one mitotic figure per high power field. Necrosis was evident. In addition, there were small whorled areas composed of large tumor cells with abundant eosinophilic cytoplasm, consistent with squamous morules (Figure 2).
Figure 1. On histologic section, the tumor was mainly
composed of neoplastic cells in acini-like structures
(H&E, x100).
Figure 2. A squamous morule (H&E, x200).
Immunohistochemistry showed the tumor cells to be positive for pancytokeratin, CK7, low molecular weight cytokeratins, trypsin, alpha-1 antitrypsin, alpha antichymotrypsin, and variably positive for synatophsin, chromogranin, and neuron specific enolase. The tumor cells were negative for insulin, glucagons, gastrin, and VIP. There was focal weak staining for somatostatin, CD99, and progesterone receptors. The findings of squamous morules and tumor cells resembling pancreatic acini on histology, together with the presence of epithelial, endocrine, and exocrine cells on immunohistochemistry confirmed a diagnosis of metastatic pancreatoblastoma.
The final pathology reports of the biopsies of the pancreatic mass - in contrast to the previous frozen sections - showed similar histologic features characteristic of pancreatoblastoma, making it the presumed primary lesion. Two and one-half months after his initial operation, the patient was therefore admitted for resection of the presumed primary lesion in the head of the pancreas. A CT scan prior to the second operation showed ascites and postoperative inflammatory changes in the pancreatic bed, as well as a small pseudocyst, but still no evidence of a pancreatic tumor (Figure 3).
Figure 3. A computed tomography scan after the first
operation showed a small pseudocyst but did not reveal
evidence of a pancreatic tumor.
Repeat exploration revealed a palpable mass, measuring approximately 5x5x5 cm, in the head of the pancreas. A pancreaticoduodenectomy with distal gastrectomy was performed. A frozen section of the pancreatic margin was negative for malignant cells. No lymph node involvement was noted. In the final pathology report following surgery, the tumor histology and immunohistochemistry was consistent with the findings of the first resection. Tumor was seen infiltrating the main pancreatic duct. Of note, despite the negative margins on frozen sections, microscopic tumor cells were evident at the margin of resection on one slide.
Following surgery, the patient was treated with adjuvant radiation and cisplatin and doxorubicin (PLADO) chemotherapy. During the next few years, the patient developed complications of his medical treatment, including bleeding at his gastrojejunostomy anastomosis that was within the radiation field and bilateral osteonecrosis of the femoral heads, both of which were treated surgically. The patient today remains free of recurrent disease 60 months after his initial operation and 57 months after his pancreaticoduodenectomy. Repeat CT scans have also shown no signs of recurrent disease. He has had normal liver function tests and normal levels of CA 125, CA 19-9 and CEA to date.
DISCUSSION
Adult pancreatoblastoma is an extremely rare neoplasm with optimal treatment regimens still uncertain. Although slightly over 200 cases have been described in the world literature, most involve patients less than 10 years old [1]. Adult cases present with slightly different clinical characteristics and outcomes than those in children. We defined adult cases as those occurring in patients 19 years of age or older, according to the criteria described by Klimstra et al. in 1995 [3]. Since 1986, when Palosaari et al. presented the first case of adult pancreatoblastoma in a 37-yearold 2], 15 additional case reports have been published, which are reviewed in Table 1 along with our patient and Palosaari’s. There are also nine other cases reported in the literature (mostly in reviews or described in the pathology and radiology literature) for which details of the cases and patient outcomes were not available [4, 5, 6, 7].
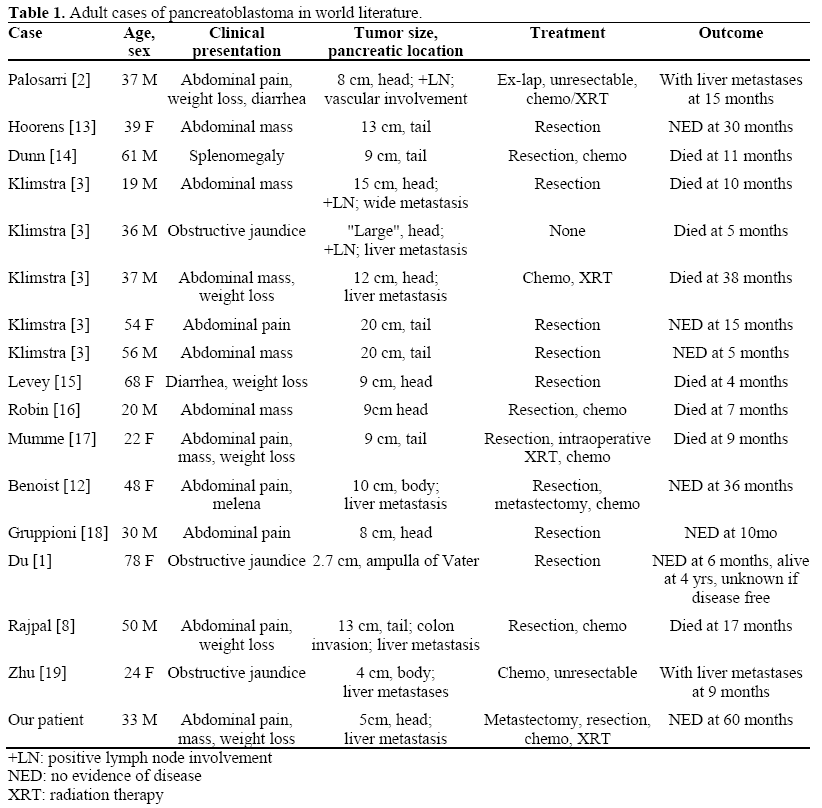
Adults with pancreatoblastoma seem to have a generally poorer prognosis than children [3]. Six of 17 cases died within one year of diagnosis (Table 1). The survival difference between pediatric and adult cases may reflect different tumor biology. One clue is that pediatric cases frequently have elevated serum tumor markers. Alpha-fetoprotein is often high in children with pancreatoblastoma but has not been consistently elevated in adults [8]. Whereas children can sometimes have prolonged survival with chemotherapy and radiation without surgical resection, surgery seems to offer the only chance of long-term survival in adults. Details of the etiology, presentation, and management of adult pancreatoblastoma are described below.
Genetics
Unlike pancreatic ductal adenocarcinoma, pancreatoblastoma does not seem to exhibit kras oncogene or p53 tumor suppressor mutations. Abraham’s group found frequent mutations of APC/B-catenin pathway chromosome 11p leading them to postulate a genetic kinship to hepatoblastoma [9]. Due to the small number of adult cases, it is unclear whether there is a genetic difference between pediatric and adult pancreatoblastoma. Several pediatric cases of pancreatoblastoma occurred in patients with Beckwith- Wiedemann syndrome and one case occurred in a patient with familial adenomatosis polyposis [9].
Tumor Location
The pancreatic head was the most common site of tumor origin, found in 8 of 17 adults (47%), followed by the tail in 6 (35%), the body in 2 (12%), and the ampulla in 1 (6%) (Table 1). This is comparable to the review of Dhebri et al. of both pediatric and adult patients [10]. The liver is the most common site of metastasis.
Histopathology
The distinctive pathologic features of pancreatoblastoma are the combined features of ductal, acinar, and endocrine cells both histologically and immunohistochemically, and the presence of squamoid corpuscles. Grossly, the tumors tend to be large at the time of diagnosis, often with metastases.
Symptoms
Most adult patients present with abdominal pain or a mass (Table 1). Weight loss, anorexia, change in bowel habits, and jaundice are other common symptoms on initial presentation. Patients may also manifest symptoms of endocrine abnormalities. One pediatric patient presented with Cushing’s syndrome as the result of an ACTH-secreting tumor [11]. Our patient presented with a tender right upper quadrant mass due to a large liver metastasis.
Laboratory
In pediatric cases, serum CEA and AFP are often elevated whereas adult pancreatoblastomas seem not to secrete AFP, CA 19-9, chromogranin A, or CEA. Frequently, as in our patient, tumor markers and liver function tests are unremarkable. Anemia is sometimes reported [8]. In only one case, Du et al. reported an elevated preoperative CA19-9 in an adult with pancreatoblastoma [1]. Rajpal et al. reported a case with an elevated lipase and proinsulin level [8]. Occasionally, adult pancreatoblastoma causes liver function test abnormalities due to biliary obstruction or hepatic metastasis.
Imaging
Most tumors are well-defined on crosssectional imaging. However, in the report by Montemarano et al. [4], only five of ten patients (9 children, 1 adult) had their tumors identified as pancreatic in origin on preoperative radiologic studies [4]. Although pancreatoblastomas often arise in the pancreatic head, biliary obstruction is uncommon. With magnetic resonance imaging (MRI), the tumors are most often described as heterogeneous with lowintermediate T1 signal intensity and high T2 intensity. Enhancement on CT is common [4].
Treatment
In the adult population, surgical resection remains the mainstay of treatment and appears to offer the only chance of cure. The three of 16 patients who did not undergo resection all died of their disease. Interestingly, Klimstra et al. reported a relatively long survival with radiation and chemotherapy alone in one patient [3]. Although the patient eventually died of his disease at 38 months, it demonstrates that radiation and chemotherapy can have a role in management. Metastatic lesions should also be resected if possible. Benoist et al. first reported a patient with successful resection of two hepatic lesions along with the primary tumor in the pancreatic body [12]. Our patient is the second reported case of resection of both the primary mass and metastasis. Both Benoist’s patient and ours achieved long-term diseasefree survival of 36 and 60 months, respectively.
SUMMARY
The long-term survival observed in our patient is remarkable considering the finding of microscopic malignant cells at the margin of resection on pathology. Despite this, after receiving radiation and chemotherapy, our patient remains disease-free five years after resection, even by subsequent exploratory laparotomy. To our knowledge, he has the longest disease-free survival in an adult reported in the world literature. Aggressive surgical resection of the primary tumor and metastatic liver lesions should be the mainstay of therapy for pancreatoblastoma. The goal of surgical resection should be disease-free margins. The role of adjuvant radiation and chemotherapy when microscopic disease is found at the surgical margin is uncertain but certainly warrants further study.
Acknowledgement
The authors would like to acknowledge the assistance of Michael Kluger, MD. This case was presented at the 2006 AHPBA Meeting, Miami Beach, FL, USA
Conflict of interest and funding
None
References
- Du E, Katz M, Weidner N, Yoder S, Moossa AR, Shabaik A. Ampullarypancreatoblastoma in an elderly patient: a case report and review of the literature. Arch Pathol Lab Med 2003; 127:1501-15. [PMID 14567752]
- Palosaari D, Clayton F, Seaman J. Pancreatoblastoma in an adult. Arch Pathol Lab Med 1986; 110:650-2. [PMID 3013120]
- Klimstra DS, Wenig BM, Adair CF, Heffess CS. Pancreatoblastoma: a clinicopathologic study and review of the literature. Am J Surg Pathol 1995; 19:1371-89. [PMID 7503360]
- Montemarano H, Lonergan GJ, Bulas DI, Selby DM. Pancreatoblastoma: Imaging findings in 10 patients and review of the literature. Radiol 2000; 214:476-82. [PMID 10671596]
- Rosebrook JL, Glickman JN, Mortele KJ. Pancreatoblastoma in an adult woman: sonography, CT, and dynamic gadolinium-enhanced MRI features. AJR Am J Roentgenol 2005; 184(3 Suppl):S78-81. [PMID 15728031]
- Cao L, Liu D. Diagnosis and Treatment of Pancreatoblastoma in China. Pancreas 2007; 34:92-5. [PMID 17198189]
- Hayasaki N, Miyake N, Takahashi H, Nakamura E, Yamagishi S, Kuno Y, et al. A case of pancreatoblastoma in an adult. Japanese Journal of Gastroenterology 1999; 96:558-63. [PMID 10369002]
- Rajpal S, Warren RS, Alexander M, Yeh BM, Grenert JP, Hintzen S, et al. Pancreatoblastoma in an adult: case report and review of the literature. J Gastrointest Surg 2006; 10:829-36. [PMID 16769539]
- Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee J, Yeo CJ, et al. Distinctive molecular genetic alterations in sporadic and Familial Adenomatous Polyposis-associated pancreatoblastomas: frequent alterations in the APC/B-catenin pathway and chromosome 11p. Am J Pathol 2001; 159:1619-27. [PMID 11696422]
- Dhebri AR, Connor S, Campbell F, Ghaneh P, Sutton R, Neoptolemos JP. Diagnosis, treatment and outcome of pancreatoblastoma. Pancreatology 2004; 4:441-53. [PMID 15256806]
- Kletter GB, Sweetser DA, Wallace SF, Sawin RS, Rutledge JC, Geyer JR. ACTH-secreting pancreatoblastoma: a case report and review of the literature. J Pediatr Endocrinol Metab 2007; 20:639-42. [PMID 17642425]
- Benoist S, Penna C, Julié C, Malafosse R, Rougier P, Nordlinger B. Prolonged survival after resection of pancreatoblastoma and synchronous liver metastases in an adult. Hepatogastroenterology 2001; 48:1340-2. [PMID 11677959]
- Hoorens A, Gebhard F, Kraft K, Lemoine NR, Kloppel G. Pancreatoblastoma in an adult: it separation from acinar cell carcinoma. Virchows Arch 1994; 424:485-90. [PMID 8032529]
- Dunn JL, Longnecker DS. Pancreatoblastoma in an older adult. Arch Pathol Lab Med 1995; 119:547- 51. [PMID 7605173]
- Levey JM, Banner BF. Adult pancreatoblastoma: a case report and review of the literature. Am J Gastroenterol 1996; 91:1841-4. [PMID 8792711]
- Robin E, Terris B, Valverde A, Molas G, Belghiti J, Bernades P, Ruszniewski P. Pancreatoblastoma in adults. Gastroentero Clin Biol 1997; 21:880-3. [PMID 9587540]
- Mumme T, Büttner R, Peiper C, Schumpelick V. Pancreatoblastoma: a rare malignant neoplasm in early adulthood. Chirurg 2001; 72:806-11. [PMID 11490758]
- Gruppioni F, Casadei R, Fusco F, Calculli L, Marrano D, Gavelli G. Adult pancreatoblastoma. A case report. Radiol Med 2002; 103:119-22. [PMID 11859308]
- Zhu L, Sidhu G, Cassai N, Yang G. Fine-needle aspiration cytology of pancreatoblastoma in a young woman: report of a case and review of the literature. Diagn Cytopathol 2005; 33:258-62. [PMID 16138370]




