Keywords
Tooth agenesis; Mutations; Hypodontia; Oligodontia; Polymorphisms
Introduction
Tooth agenesis is genetically and phenotypically a heterogeneous condition. It is assumed that different phenotypic forms are caused by different genes involving different interacting molecular pathways, providing an explanation not only for the wide variety in agenesis patterns but also for associations of dental agenesis with other oral anomalies. Tooth agenesis of one or more teeth comprises one of the most common cranio-facial anomalies in human. It occurs either as an isolated/familial trait or in association with various genetic syndromes. It is transmitted as an autosomal dominant, recessive or X-linked trait. In addition to aesthetic significance, it may affect activities such as mastication, speech alteration and malocclusion. It has been reported in literature that the frequency of missing teeth excluding third molar varies from 2% to 11% in different populations [1]. However, missing wisdom teeth has been reported in 1/5 of the population. Permanent teeth that are found missing include second premolar as most common (3.5%) closely followed by maxillary lateral (1.8%) incisors [2,3]. However, a significant variability with regard to location, symmetry, number of teeth involved, size, shape and developmental stage has been extensively reported in different populations. In most cases, the condition has a genetic basis; however it may also be caused by other factors that include trauma, chemotherapy and radiation therapy and use of thalidomide during pregnancy [4].
Genes Involved in Tooth Development and Differentiation
The molecular aspects of tooth development are similar to those in the development of other organs that include epithelial-mesenchymal interactions. Nearly 300 groups of molecules are involved in tooth development such as fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), sonic hedgehog (SHH), and wingless integrated (WNTs). As mentioned previously, BMP4 acts antagonistically with FGF8 and this interaction plays a role in the periodic patterning [5]. The molecular dialogue between oral ectoderm and odontogenic mesenchyme during tooth development is very complicated (Figure 1) [6]. There is a vast knowledge about the signal interchanges that are crucial to control differentiation and morphological changes and spatiotemporal expression of specific genes during teeth formation, yet little is known about their up- or down-regulation [7]. In vitro and in vivo studies have identified that both MSX1 and PAX9 are essential for the induction of bone morphogenetic protein 4 (BMP4), which is considered to be a pivotal step in tooth development in mammals [8]. Evidence so far points to the direction that there is no single gene directly connected with ontogenesis or the lack of any specific tooth. Instead, tooth initiation and morphogenesis occur by an orchestration of numerous genetic and epigenetic factors [6,9]. At the same time, most of the developmental defects in teeth usually occur as a result of mutations in genes encoding signaling molecules and transcription factors [9] such as mutations in the PAX9 gene resulting in partial or total anodontia (missing all teeth, either primary and/or permanent) and mutations in Runt-related transcription factor 2 (RUNX2) causing supernumerary teeth [10-12]. Aim of this review is to present the information associated with the mutations in genes associated with tooth agenesis, therefore, no more information related with the genes involved in tooth development and differentiation is presented in this review.
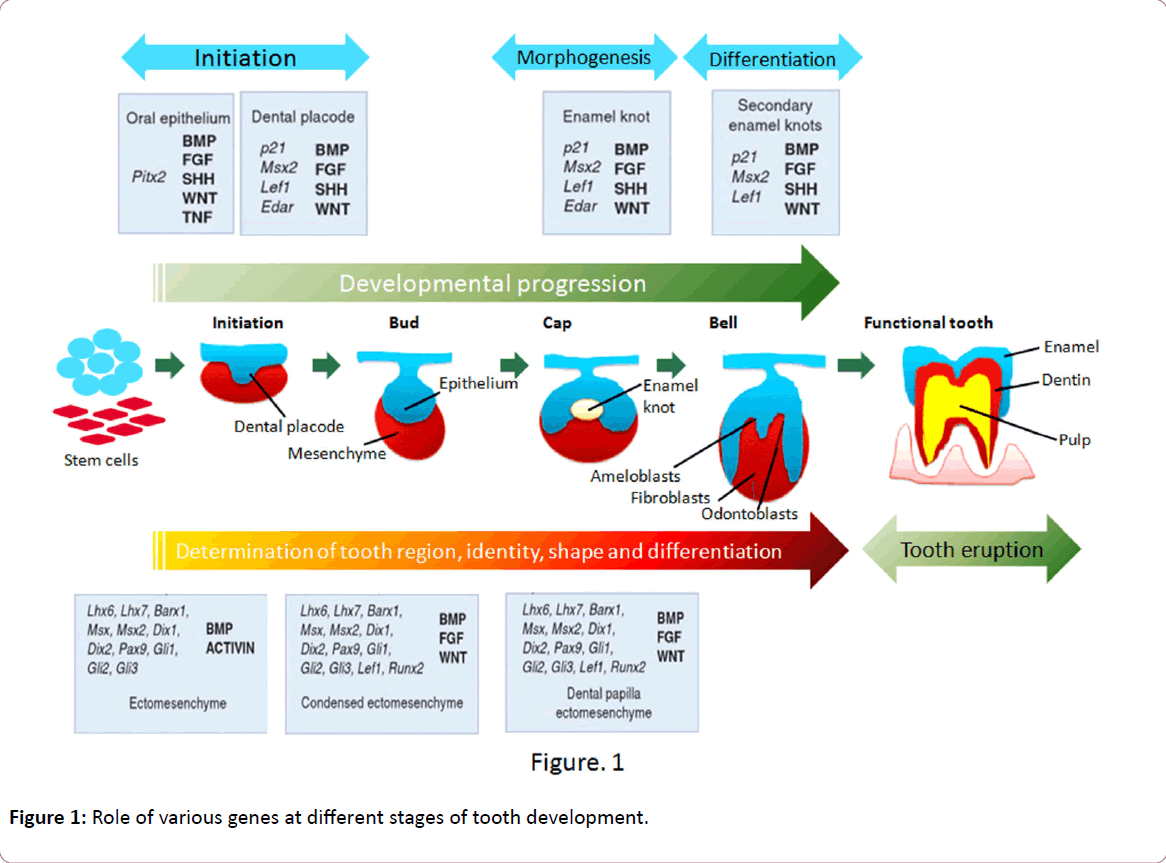
Figure 1: Role of various genes at different stages of tooth development.
The development of tooth is primarily controlled genetically [13]. More than 200 genes have been found to be expressed during this process [14]. Most often reported genes associated with non-syndromic tooth agenesis are PAX9, MSX1, EDA, and AXIN2 [15-20]. Mutations of these genes have an impact on agenesis patterns, although in notably different ways. PAX9 sequence alterations lead to agenesis mainly of molars [15,21] and MSX1 mutations have been implicated primarily in congenitally missing premolars [22,23]. Severe agenesis of both molars and premolars has been noted in patients with an AXIN2 mutation [24] whereas missing incisors are the chief manifestation of EDA-associated non-syndromic oligodontia [20,25-27].
MSX1 Gene
MSX1 gene initially named as homeobox 7 (HOX7) is a nonclustered homeobox protein located on the chromosome 4p16.3-p16.1 [28]. This gene encodes a member of the muscle segment homeobox gene family (Figure 2). The encoded protein functions as a transcriptional repressor during embryogenesis through interactions with components of the core transcription complex and other homeoproteins [29]. It has been shown to have a significant role in tooth development [30]. Mice with a homozygous deletion in Msx1 exhibited a complete secondary cleft palate, complete failure of incisor development, and budstage arrest of molar development [31]. MSX1 was the first gene to be evidently associated with human tooth agenesis [32]. Mutations in this gene have generally been associated with autosomal dominant inheritance of hypodontia (missing up to five permanent teeth, excluding the 3rd molars) as well as oligodontia (missing six or more permanent teeth, excluding 3rd molars or wisdom teeth) [32]. However, in 2006, first association of MSX1 mutations with an autosomal recessive type of hypodontia was reported from Pakistan [17]. Mutations in MSX1 such as p.G22RfsX168 [22], p.M61K [30], p.R176W [33], p.Q187X [16], p.Q189X [34], p.A194V [35], p.R196P [32], p.Q216QfsX125 [33], p.A219T [17], p.A221E [36] has been found to be associated with non-syndromic tooth agenesis. Deletion of chromosome 4p [37] and mutations in MSX1 gene such as p.S104X [38] and p.S202X [39] have also been reported to produce tooth agenesis associated with other symptoms. Recently a novel mutation has been reported in this gene (c. 910_911dupTA; p.*304Tyrext*48) disrupting the original stop codon [40]. Insertion such as 750_751insACCGGCTGCC p.F251Pfsx92 and c.452_9G>A c.451_452insCCCTCAG (7bp ins) p.R151FsX20 [41] have also been recently reported to associated with tooth agenesis. Mutations in MSX1 gene that are associated with tooth agenesis are summed up in Table 1.
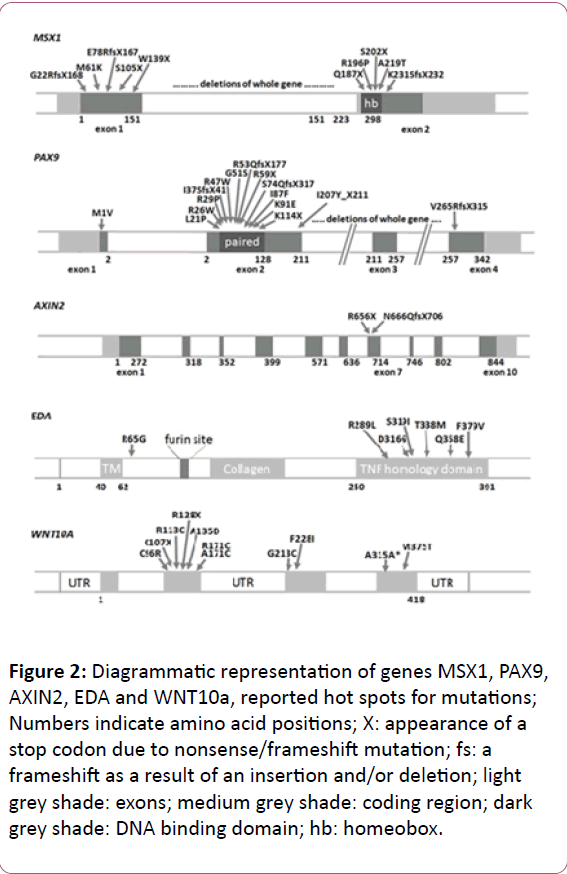
Figure 2: Diagrammatic representation of genes MSX1, PAX9, AXIN2, EDA and WNT10a, reported hot spots for mutations; Numbers indicate amino acid positions; X: appearance of a stop codon due to nonsense/frameshift mutation; fs: a frameshift as a result of an insertion and/or deletion; light grey shade: exons; medium grey shade: coding region; dark grey shade: DNA binding domain; hb: homeobox.
| Phenotype |
Mutation |
Type |
Reference |
| Oligodontia |
*6C>T Polymorphism |
Polymorphism |
42 |
| Oligodontia |
c.62dupG (p.G22RfsX168) |
Frameshift Mutation |
22 |
| Hypodontia |
5354C > G; A40G |
Missense mutation |
43 |
| Oligodontia |
c.182T>A (p.M61K) |
Missense Mutation |
30 |
| Oligodontia |
c.314C>A (p.S104X) |
Nonsense Mutation |
38 |
| Hypodontia |
c.347C>T (G116G) |
Missense mutation |
43 |
| Oligodontia |
c.416G>A (p.W139X) |
Nonsense Mutation |
44 |
| Hypodontia |
c.453G>T (p.R151S) |
Missense mutation |
45 |
| Oligodontia |
c.452_9G>A,c.451_452insCCCTCAG(7bp ins) (p.R151FsX20) |
Nonsense mutation |
41 |
| Hypodontia |
c.463C>A (p.P155Q) |
Missense mutation |
43 |
| Hypodontia |
c.517C>A (p.R173S) |
Missense mutation |
46 |
| Oligodontia |
c.526C>T (p.R176W) |
Missense mutation |
33 |
| Oligodontia |
c.559C>T (p.Q187X) |
Nonsense Mutation |
16 |
| Hypodontia |
c.572_573ins GCAAGTT: p.F191fs |
Frameshift insertion |
34 |
| Oligodontia |
c.581C>T (p.A194V) |
Missense Mutation |
35 |
| Oligodontia |
c.605C>A (p.S202X) |
Nonsense Mutation |
39 |
| Oligodontia |
c.614T>G (p.L205R) |
Missense mutation |
46 |
| Oligodontia |
c.644insA (p.Q216QfsX125) |
Frameshift mutation |
33 |
| Oligodontia |
c.665G>A (p.A219T) |
Missense Mutation |
17 |
| Oligodontia |
c.662C>A (p.A221E) |
Missense mutation |
36 |
| Oligodontia |
c.665_666insA (p.N222KFsX118) |
Frameshift Mutation |
47 |
| Hypodontia |
c.671 T>C (p.L224P) |
Missense Mutation |
48 |
| Oligodontia |
c.707delG (p.K237SFsx2), |
Frameshift Mutation |
47 |
| Hypodontia |
750_751insACCGGCTGCCp.F251Pfsx92 |
Frameshift mutation |
49 |
| Oligodontia |
c.910_911dupTA (p.*304Yext*48) |
Nonsense Mutation |
40 |
Table 1: Mutations detected in MSX1 gene associated with tooth agenesis.
PAX9 Gene
PAX9 is a member of the paired box (PAX; Figure 2) family of transcription factors and is located on chromosome 14q12-q13 [50]. It plays an integral role both during foetal development and cancer growth [51]. It is evident that this gene also plays important roles during tooth development, as shown by its expression pattern, the phenotype of transgenic mice lacking both copies of the gene, and by the association of agenesis of posterior dentition with PAX9 mutations in humans.10 [52]. Probably it mediates its tooth-specific functions through its interactions with other similar proteins. It has been shown that an important partnership between the PAX9 paired domain protein and the MSX1 homeoprotein is responsible for regulating gene expression in dental Mesenchyme [53].
PAX9 gene was first shown to be associated with all forms of the disease including autosomal dominant, non-syndromic and familial oligodontia [52]. Since then several novel mutations in the gene have been discovered in familial and non-familial forms of the disease. However, a more recent study has found families with affected benign hereditary chorea also showing deletion in PAX9 gene resulting in oligodontia [53]. Mutations reported so far range from being missense that changes just one amino acid in the entire protein to premature stop codons that result in truncation of the protein products. It is also possible that the mutant allele may be present in its hypomorphic state thus indicating the combined activities of both its wild and mutant alleles do not reach the threshold level required for normal tooth development. However, it is also possible that relatively milder phenotypes may result due to defective allele that generates an aberrant protein which acts in a dominantnegative manner or perhaps has a novel function [21,54]. Only a few reports exist in the literature that has been found to be associated with agenesis of deciduous teeth with PAX9 mutations [21,55,56]. Whole gene deletions and the start codon mutations in PAX9 gene caused the most severe teeth developmental defects extending to the premolars and primary molars, i.e. affecting the whole postcanine dentition [55,56]. It also establishes the time and place of morphogenesis [22]. Mutations detected so far in PAX9 gene are summed up in Table 2.
| Phenotype |
Mutation |
Type |
Reference |
| Oligodontia |
c.1A?G (p.M1V) |
Missense Mutations |
56 |
| Hypodontia |
c.16G?A (p.G6R) |
Missense Mutations |
57 |
| Hypodontia |
c.43T>A (p.F15I) |
Missense Mutation |
58 |
| Hypodontia |
c.59delC (p.P20Rfs*65) |
Frameshift Mutation |
59 |
| Oligodontia |
c.62T?C (p.L21P) |
Missense Mutations |
15 |
| Oligodontia |
c.76C?T (p.R26W) |
Missense Mutations |
60 |
| Oligodontia |
(p.L27P) |
Missense Mutations |
61 |
| Oligodontia |
c.83G?C (p.R28P) |
Missense Mutations |
62 |
| Oligodontia |
(p.I29T) |
Missense Mutations |
61 |
| Oligodontia |
c.109-110insG (p.I37Sfs*41) |
Frameshift Mutation |
63 |
| Hypodontia |
c.128G?A (p.S43K) |
Missense Mutations |
57 |
| Oligodontia |
c.139C?T (p.R47W) |
Missense Mutations |
63,64 |
| Oligodontia |
c.140G?C (p.R47P) |
Missense Mutation |
47 |
| Oligodontia |
c.151G?A (p.G51S) |
Missense Mutations |
65 |
| Oligodontia |
c.167C?T (p.I56T) |
Missense Mutation |
47 |
| Oligodontia |
c.175C?T (p.R59*) |
Nonsense Mutations |
66 |
| Oligodontia |
c.175-176ins288 (p.R59Zfs*177) |
Nonsense Mutations |
15 |
| Oligodontia |
c.597-599dup (p.D200Sfs*13) |
Nonsense Mutations |
52 |
| Oligodontia |
c.259A?T (p.I87F) |
Missense Mutations |
67 |
| Oligodontia |
c.271A?G (p.K91E) |
Missense Mutations |
15 |
| Oligodontia |
c.321_322insG |
Frameshift mutation |
68 |
| Oligodontia |
c.340A?T (p.K114*) |
Nonsense Mutations |
21 |
| Oligodontia |
c.433C?T (p.Q145*) |
Nonsense Mutations |
18 |
| Hypodontia |
c.73-75 delATC (p.C146T) |
Missense mutation |
69 |
| Hypodontia |
c.480C>G (p.Y160X) |
Nonsense Mutation |
70 |
| Oligodontia |
c.503C?G (p.A168G) |
Missense mutations |
71 |
| Oligodontia |
c.619-621 del ATCins24bp(p.I207Yfs*211) |
Frameshift Mutation |
67 |
| Oligodontia |
c.718G>C (p.A240P) |
Missense Mutation |
72 |
| Oligodontia |
c.792-793insC (p.V265Rfs*315) |
Frameshift Mutation |
73 |
| Hypodontia |
g.718C (C Allele), C912T (T Allele),A1031G (G allele) |
Polymorphism |
74 |
| Oligodontia |
g.10672A > T (p.D40I) |
Missense Mutation |
43 |
Table 2: Mutations in PAX9 gene associated with tooth agenesis.
AXIN2 Gene
AXIN2 or axis inhibitor protein 2 is a gene located on chromosome 17q23-q24 [75]. It is a signaling molecule in the ‘wingless-type MMTV integration site family’ (WNT) pathway that starts operating very early in tooth placode formation (Figure 2). The association of the gene to tooth agenesis was first found in a Finnish family with a predisposition for colorectal cancer [24]. It has been shown that the AXIN2 mutations may also be responsible for sporadic forms of incisor agenesis [76]. Mutations in human AXIN2 associated with oligodontia have been reported [24,33,77]. In humans, mutations in AXIN2 cause tooth agenesis that affects permanent teeth predominantly, including the permanent molars, lower incisors, and upper lateral incisors. AXIN2 is involved in sporadic forms of common incisor agenesis [78,79]. Mutations detected so far in AXIN2 gene are summed up in Table 3. LTBP3 (latent transforming growth factor beta binding protein 3) is a gene that modulates the bioavailability of Transforming growth factor beta 1 (TGF-β) [80,81]. This gene is located on chromosome 11q12 [82]. This gene has been found to be the causal factor in a Pakistani family showing autosomal recessive form of familial oligodontia [83].
| Phenotype |
Mutation |
Type |
Reference |
| Oligodontia |
c.923C>T (p.T308M) |
Missense Mutation |
78 |
| Hypodontia |
c.1387 C>T (p.R463C) |
Missense mutation |
79 |
| Oligodontia |
c.C1978T (p.H660Y),c.1966C>T (p.R656Stop) and c.1994delG (p.L688Stop) |
Missense mutation
Nonsense Mutation |
24 |
| Oligodontia |
c.1989G>A (p.W663X) |
Nonsense mutation |
80 |
| Oligidontia |
c.1999dupG (p.G666GfsX42)
c.2051C>T (p.A684V)
c.2272G>A (p.A758T) |
Frameshift Mutation
Missense mutation
Missense mutation |
33 |
| Oligodontia |
c.2490G>C (p.M830I) |
Missense Mutation |
78 |
Table 3: Mutations found in AXIN2 gene related with tooth agenesis.
EDA1 Gene
EDA1 (ectodysplasin 1) is located on chromosome Xq12-q13.1 (Figure 2) that encodes a protein which belongs to the tumor necrosis factor (TNF) superfamily of ligands. In total, eight isoforms of the EDA transcript can be created by differential splicing of the 12 exons. It has been associated to X-linked recessive form of ectodermal dysplasia [84]. A study of Chinese families with non syndromic X-linked hypodontia showed that a Thr338Met mutation of the EDA gene was responsible for the congenital absence of maxillary and mandibular central incisors, lateral incisors, and canines, with the high possibility of persistence of maxillary and mandibular first permanent molars [26]. Both Ectodysplasin A receptor (EDAR) and Ectodysplasin-A receptor-associated adapter protein (EDARADD) encode respectively the proteins EDAR and EDARADD that belongs to EDA/EDAR/NF-κβ signaling pathway. EDA-EDAR (complex formed by EDA and its receptor) binds to the downstream adaptor EDARADD which leads to activation of the transcription factor NF-κβ (nuclear factor NF-κB pathway). It is essential for the development of hair follicles, teeth, exocrine glands and other ectodermal derivatives [85,86]. Evidence so far also points to the direction that sometime heterozygous mutations in EDA are present in addition to mutations in Wnt Family Member 10A (WNT10A) as digenic mutations [87]. More than seventy mutations in EDA gene have been identified and more than 2/3 of these are reported in conserved TNF homology domain. Only thirteen have been found to be linked with non-syndromic tooth agenesis [88-91]. Of these, five residues (Gln331, Met364, Val365, Ser374 and Ser319) can form a cluster with slightly negative surface potential and play critical role during receptor binding activity of EDA and several other Tumour necrosis factorlike (TNF) domain containing protein [92]. In 2008, a Chinese study reported p.Asp316Gly mutation in EDA protein to be associated with non-syndromic congenital tooth agenesis affecting every tooth type. All affected members show differential expression [93]. Mutations detected so far in EDA and related genes are summed up in Tables 4-6.
| Phenotype |
Mutation |
Type |
Reference |
| Hypodontia |
c.193C>G, (p.R65G) |
Missensemutation |
94 |
| Oligodontia |
c.252DelT (p.P84PfsX6, c.457C>T(p.R153C), c.466C>T (p.R156C) |
Frameshift Mutation
Missense mutation |
95 |
| Oligodontia |
c.612-29del18bp (IPGIPG205-210del) |
Frameshift Mutation |
47 |
| Oligodontia |
c.769G>C (p.G257R) |
Missense mutation |
95 |
| Oligodontia |
c.776C>A (p.A259E) |
Missense mutation |
27 |
| Hypodontia |
(c.779 T>G) (p.I260S) |
Missense mutation |
88 |
| Oligodontia |
c.865C>T (p.R289C) |
Missense mutation |
27 |
| Oligodontia |
c.866G>T (p.R289L) |
Missense mutation |
91 |
| Hypodontia |
c.874G>T (p.E292X) |
Nonsense mutation |
96 |
| Oligodontia |
c.901-904delTACT (p.T301SfsX5) |
Nonsense mutation |
47 |
| Oligodontia |
c.936C>G (p. I312M) |
Missense mutation |
95 |
| Hypodontia |
c.947A>G (p.D316G) |
Missense mutation |
93 |
| Oligodontia |
c.956G>T (p.S319I) |
Missense mutation |
3 |
| Hypodontia |
c.993G>C (p.Q331H) |
Missense mutation |
25 |
| Oligodontia |
c.1001G>A (p.R334H) |
Missense mutation |
27 |
| Hypodontia |
c.1013 C>T (p.T338M) |
Missense mutation |
93 |
| Oligodontia |
c.1045G>A (p.A349T) |
Missense mutation |
95 |
| Oligodontia |
c.1069C>T (p.R357W) |
Missense mutation |
47 |
| Hypodontia |
c.1071G>A (p.Q358E) |
Missense Mutation |
90 |
| Hypodontia |
c.1091T>C (p.M364T) |
Missense mutation |
97 |
| Hypodontia |
c.1336T>C (p.V365A) |
Missense mutation |
89 |
| Oligodontia |
c.1133C>T (p.T378M) |
Missense mutation |
47 |
| Oligodontia |
c.1135T>G (p.F379V) |
Missense mutation |
91 |
Table 4: Mutations found in EDA gene associated with tooth agenesis.
| Phenotype |
Mutation |
Type |
Reference |
| Oligodontia |
c.43G>A (p.V15I); c.319A>G (p.M107V), c.871G>A (p.A291T) |
Missense Mutations |
95 |
| Oligodontia |
c.973C>T (p.R325W); c.1073G>A (p.R358Q) |
Missense Mutations |
47 |
| Oligodontia |
c.1072C > T (p. R358X) |
Nonsense Mutation |
98 |
| Oligodontia |
c.1109T>C (p.V370A) |
Missense Mutations |
95 |
| Oligodontia |
c.1135G>A (p.E379K) |
Missense Mutation |
47 |
| Oligodontia |
c.1138A>C (p.S380R) |
Missense Mutation |
95 |
| Oligodontia |
c.1172T>A (p.M391K) |
Missense Mutation |
47 |
Table 5: Mutations found in EDAR gene associated with tooth agenesis.
| Phenotype |
Mutation |
Type |
Reference |
| Oligodontia |
c.27G>A (p.M9I) |
Missense Mutation |
95 |
| Oligodontia |
c.308C>T(p.S103F), c.508C>T (p.R170W) |
Missense Mutation |
47 |
Table 6: Mutations found in EDARADD gene associated with tooth agenesis.
WNT10A Gene
WNT10A located adjacent to another gene of the same family Wnt Family Member 6 (WNT6) at 2q35 in humans is active during the development of hair follicles and limbs (Figure 2), and in haematopoiesis. It has been found to be expressed in many adult tissues such as lymph nodes, blood, adrenal gland, prostate, testis, ovary, retina, brain, lung, kidney etc. and may also play an important role in many cancers. Functional studies showed that WNT10A activates the canonical WNT pathway and regulates mesenchymal cell fate in that it inhibits adipogenesis and stimulates osteoblastogenesis [99]. The canonical WNT-β catenin pathway is essential for tooth morphogenesis of both primary and secondary dentition [100]. A recent study in mice has shown that inhibition of the canonical WNT signalling arrests tooth development at early stages [101] whereas constitutive activation of epithelial WNT/β catenin signalling leads to continuous tooth generation [102]. WNT10A is a key mediator of WNT signaling in tooth morphogenesis and, in line with this, WNT10A has been found to be associated with inherited tooth agenesis [103,104]. Van den Boogaard and co-workers have shown that more than half of the patients with non-syndromic oligodontia carry either bi-allelic or mono-allelic mutations in WNT10A [105,106]. In addition to this, WNT10A mutations have also been found in a large proportion of patients having oligodontia and mild manifestations of ectodermal dysplasia.106 WNT/β-catenin signaling has been also reported to play an important role in establishing the size and morphology of the adult tooth [89]. Mutations detected in WNT10a gene are summed up in Table 7.
| Phenotype |
Mutation |
Type |
Reference |
| Hypodontia |
21-bp deletion combined with
1-bp insertion, c.-14_7delinsC |
Frameshift Mutation |
107 |
| Oligodontia |
c.208C>T (p.R70W) |
Missense Mutation |
47 |
| Hypodontia |
c.321C> A (p.C107*) |
Nonsense Mutation |
105 |
| Oligodontia |
c.337C>T (p.R113C),
c.433G>A (p.V145M),
c.433G>T (p.V145L),
c.460C>A (p.L154M),
c.493G>A (p.G165R) |
Missense Mutation |
47 |
| Oligodontia |
c.511C>T (p.R171C) |
Missense Mutation |
89,95 |
| Oligodontia |
c.579-592del (p.E194AfsX28)
GGAACACCCAGCCC |
Frameshift Mutation |
47 |
| Oligodontia |
c.637G>A (p.G213S) |
Missense Mutation |
95 |
| Oligodontia |
c.664G>T (p.E222X) |
Nonsense Mutation |
47 |
| Hypodontia |
c.682T>A (p.F228I) |
Missense Mutation |
47,105 |
Table 7: Mutations in other genes including WNT10A associated with tooth agenesis.
The information available in the literature with regard to reported mutations such as PAX9 (A240P) has been found to be polymorphic in nature at least in sporadic oligodontia in African, American, and European subpopulations. However, a recent study [108] could not establish the presence/absence of a genotype-phenotype association in two Mexican families. The inconsistency of these results might be explained to some extent by population diversities that may cause variation, as genetic polymorphisms often show ethnic disparity. On the other hand, this variation between the high prevalence of tooth agenesis and relatively small number of findings points to the direction that tooth agenesis may be a heterogeneous trait. Combination of two or more gene polymorphisms could result changes in gene structures, gene-gene and protein-protein interactions as well as a reduction in gene dosages, thereby leading to specific phenotypes. Evidence from animal studies suggests that a single homozygous mutation either in MSX1 or PAX9 gene exhibit early arrest of tooth development [51,109]. Contrary to human findings, studies in mice having a heterozygous Pax9 or heterozygous Msx1 loss-of-function mutation do not exhibit tooth developmental defects thus suggesting that different gene dosages are required for tooth development. Recent studies in mice have further strengthened this evidence that synergistic interactions are occurring during the mouse tooth morphogenesis [110]. Further to this; in vitro studies have also provided the evidence that PAX9 expression is down regulated via its interaction with MSX1 during tooth formation [111]. Therefore to further understand and broaden our knowledge about tooth agenesis, we need to perform larger case-control studies.
Conclusion and Future Directions
This review provides further insight to the orthodontists to comprehensively ask the patient about their family history of individuals having similar/dissimilar or any other dental anomalies. Observations with regard to familial and clinical history combined with molecular genetics expertise might bring about tremendous gains for better understanding of genetic causes of various tooth anomalies. Therefore, it is important that both molecular geneticists and dentists must work in tandem to further identify genes causing tooth agenesis. This information will provide enormous information not only about the underlying dental defect but also about the precise changes in the physiologic processes by the mutations in these genes. Identification of mutated genes that cause tooth agenesis (including familial, non-familial, syndromic and non-syndromic) will also enable the future studies to assess the mechanism how environmental and dietary factors modify gene expression and result in distinct but similar clinical phenotypes. Precise exposition of the pathogenic mechanism in tooth agenesis will provide further insights into the role of teeth in craniofacial development and will advance our understanding of cranioorofacial dysmorphology. Evidence gathered through human molecular genetics approach provides information that tooth agenesis is heterogeneous in nature suggesting the involvement of multigenic defects contributing to the varied clinical variability of this entity. Tooth development is a very multifaceted process and involves many genes. This review summarizes the role of mutations in genes such as MSX1, PAX9, AXIN2 etc. in causing tooth agenesis. It is still a long way to go until we finally reach our goal of better understanding the process of ontogenesis. Cross collaboration of geneticists and dentists will be reformed in this century. It is also very important to consider the involvement of epigenetic factors that can reduce gene dosage, alter genes interaction and other posttranscriptional modulation agents that also could explain dental agenesis associated or not with systemic anomalies. Emerging research areas such as human dento-orofacial genetics will become an integral part of the modern health care and will have significant impact the way we diagnose, prevent, and treat these disorders. The identification of mutations in these genes result in mutated/truncated protein and has been linked evidently with tooth agenesis. In addition to other factors, metabolic state of an individual also plays an important role in tooth development. It is anticipated that both environmental and systemic instability will result in the tooth anomalies. Therefore, it becomes quite important that diagnosis of this condition should be made precisely based on detailed account of clinical history, family history and any exposure to environmental factors such as irradiation, use of chemotherapeutic agents and dioxins. Therefore it becomes imperative to find out the actual cause of tooth agenesis in every patient.
It has been demonstrated that mutations in genes such as PAX9, MSX1, and AXIN2 etc. are associated in human tooth agenesis; it is becoming progressively more obvious that many more genes might also play an integral role in tooth development. This is undoubtedly supported by current literature classifying three specific blueprints in human tooth agenesis as: missing pattern of molars and premolars; anterior patterns involving agenesis of cuspids and/or incisors and mixed patterns with missing premolars and lateral incisors [7]. Lack of mutations in PAX9 or MSX1 in families showing a distinct pattern of posterior tooth agenesis further supports the role of so far unidentified genes in causing tooth agenesis [112]. Buccal cells are excellent source of DNA. Dental clinicians during their interactions with patient can easily collect buccal cells noninvasively using tooth brush provided the patients consent to do as well. This non-invasive approach will be more productive as it will increase the patient number ready to take part in molecular genetic studies. Therefore, more and more patients consenting to do this type of analysis will help the molecular geneticists to broaden their knowledge of tooth agenesis. Further studies in this area in different population will be helpful in further understanding the role of genes involved in tooth agenesis. It may presumably show that human dental agenesis is caused by several independent defective genes, acting alone or in combination with other genes and leading to a specific phenotypic pattern. These insights will significantly add to our knowledge of the complex cellular events that give rise to the tooth and of the molecular developmental strategies that control the patterning of the human dentition. In addition further identification of mutations in still undiscovered genes will also shed some more light to better understand this process of odontogenesis. This new knowledge will better equip the clinical dentists to diagnose and treat the patients with this developmental anomaly more accurately. Moreover, this approach will further help to personalize the dental treatment. Further advances to understand the complexity of genetic networks regulating the odontogenesis and how these gene mutations alter gene expression especially in larger cohorts will pave the way for developing an early and accurate diagnostic test to screen the affected families.
Clinical Significance
Many common genetic diseases are not inherited as single gene defect. Gene-environment interactions are important determinants. In the last decade, there is an increasing emphasis toward the evidence-based approach both in medical and dental health practice. Recent scientific advances have improved and impacted on the quality of life of many individuals. Many diseases can now be identified in its initial stages and few are diagnosed even before their onset. Hence it is quite evident that genetics plays an important role in diagnosing and treating these diseases. From dental point of view, both diet and dentists play a significant impact on one’s oral health. The new information will allow dentists to use patient’s medical history to help tailor a dental care plan specific to their needs. If one is at an increased risk of oral disease due to genetic factors, they may need special dental care treatments. People suffering from gum diseases and tooth decay may have genetic variants in some genes. However, sometimes either activation of some genes or altered expression can also result in dental anomalies. Recent developments in dental genetics don’t mean that oral health in under imminent threat. Several genetic factors can influence one’s oral health. These factors can determine the way one’s teeth are aligned and/or whether or not people will get cavities, regardless of their dental care habits. The chemical composition of saliva also determines how well it can neutralize the acids that lead to plaque formation and subsequent tooth decay. Periodontitis is also linked to genetics. Periodontal disease is one of the most common oral diseases characterized by inflammation and destruction of the periodontium. This disease results from complex gene-environment interaction. Multiple gene variants in a particular set of genes appear to confer genetic susceptibility to environment risk factors such as tobacco products, microbial infections, medical and immunological compromise, alcohol consumption, osteoporosis and variety of medications, sex and age. Hence, the treatment of oral, dental, and craniofacial diseases has drastically changed because of these new genomic advances, which are leading the researchers and clinicians to better understand oral biology. More precise and faster diagnostic tests, new drugs and biologics, practicebased research, and culturally sensitive interventions are providing novel avenues to improve oral health.
References
- Polder BJ, Van’t Hof MA, Van der, Linden FP, Kuijpers-Jagtman AM (2004) A meta-analysis of the prevalence of dental agenesis of permanent teeth. Comm Dent and Oral Epidemiol 32: 217-226.
- Symons AL, Stritzel F, Stamation J (1993) Anomalies associated with hypodontia of the permanent lateral incisor and second premolar. J ClinPediatr Dent 17: 109-111.
- Sarkar T, Bansal R, Das P (2014) Whole genome sequencing reveals novel non-synonymous mutation in ectodysplasin A (EDA) associated with non-syndromic X-linked dominant congenital tooth agenesis. PLoS One 9:e106811.
- Nieminen P (2009) Genetic basis of tooth agenesis. J ExpZool B MolDevEvol 312B: 320-342.
- Neubuser A, Peters H, Balling R, Martin GR (1997) Antagonistic interactions between FGF and BMP signaling pathways: a mechanism for positioning the sites of tooth formation. Cell 90: 247-255.
- Jernvall J, Thesleff I (2000) Reiterative signaling and patterning during mammalian tooth morphogenesis. MechDev 92: 19-29.
- Tucker A, Sharpe P (2004) The cutting-edge of mammalian development; how the embryo makes teeth. Nat Rev Genet 5: 499-508.
- Chen Y, Bei M, Woo I, Satokata I, Maas R (1996) Msx1 controls inductive signaling in mammalian tooth morphogenesis. Devel122:3035-3044.
- Koussoulakou DS, Margaritis LH, Koussoulakos SL (2009) A curriculum vitae of teeth: evolution, generation, regeneration. Int J BiolSci 5: 226-243.
- Peters H, Neubüser A, Balling R (1998) Pax genes and organogenesis: Pax9 meets tooth development. Eur J Oral Sci 106: 38-43.
- Ryoo HM, Kang HY, Lee SK, Lee KE, Kim JW (2010) RUNX2 mutations in cleidocranial dysplasia patients. Oral Dis 16: 55-60.
- Lee KE, Seymen F, Ko J, Yildirim M, Tuna EB, et al. (2013) RUNX2 mutations in cleidocranial dysplasia. Genet Mol Res 12: 4567-4574.
- Thesleff I (2003) Epithelial-mesenchymalsignalling regulating tooth morphogenesis. J Cell Sci 116: 1647-1648.
- Thesleff I, Nieminen P (2006) Tooth morphogenesis and cell differentiation. Cur Opi in Cell Bio 8: 844-850.
- Das P, Hai M, Elcock C, Leal SM, Brown DT, et al. (2003) Novel missense mutations and a 288-bp exonic insertion in PAX9 in families with autosomal dominant hypodontia. Am J of Med Genet 118A: 35-42.
- De Muynck S, Schollen E, Matthijs G, Verdonck A, Devriendt K, et al. (2004) A novel MSX1 mutation in hypodontia. Am J Med Genet A 128A: 401-403.
- Chishti MS, Muhammad D, Haider M, Ahmad W (2006) A novel missense mutation in MSX1 underlies autosomal recessive oligodontia with associated dental anomalies in Pakistani families. J of Hum Genet 51:872-888.
- Hansen L, Kreiborg S, Jarlov H, Niebuhr E, Eiberg H (2007) A novel nonsense mutation in PAX9 is associated with marked variability in number of missing teeth. Eur J of Oral Sci 115: 330-333.
- Li WL, Cui JJ, Fang QY, Mei LX (2008) [A novel mutation of MSX1 gene in a Chinese pedigree with oligodontia]. Zhonghua Kou Qiang Yi XueZaZhi 43: 157-159.
- Mues G, Tardivel A, Willen L, Kapadia H, Seaman R, et al. (2010) Functional analysis of Ectodysplasin-A mutations causing selective tooth agenesis. Eur J Hum Genet 18: 19-25.
- Nieminen P, Arte S, Tanner D, Paulin L, Alaluusua S, et al. (2001) Identification of a nonsense mutation in the PAX9 gene in molar oligodontia. Eur J Hum Genet 9: 743-746.
- Kim JW, Simmer JP, Lin BP, Hu JC (2006) Novel MSX1 frameshift causes autosomal-dominantoligodontia. J of Den Res 85:267–271.
- Vieira AR, Meira R, Modesto A, Murray JC (2004) MSX1, PAX9, and TGFA contribute to tooth agenesis in humans. J Dent Res 83: 723-727.
- Yue H, Liang J, Yang K, Hua B, Bian Z (2016) Functional analysis of a novel missense mutation in AXIN2 associated with non-syndromic tooth agenesis. Eur J Oral Sci 124: 228-233.
- Ayub M, Rehman F, Yasinzai M, Ahmad W (2010) A novel missense mutation in the ectodysplasin-A (EDA) gene underlies X-linked recessive nonsyndromichypodontia. Int J of Dermat 49: 1399-1402.
- Han D, Gong Y, Wu H, Zhang X, Yan M, et al. Novel EDA mutation resulting in X-linked non-syndromichypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur J of Med Genet 51:536-546.
- Song S, Han D, Qu H, Gong Y, Wu H, et al. (2009) EDA gene mutations underlie non-syndromicoligodontia. J Dent Res 88: 126-131.
- Campbell K, Flavin N, Ivens A, Robert B, Buckingham M, et al. (1989) The human homeobox gene HOX7 maps to 4p16.1 and is deleted in Wolf-Hirschhorn syndrome patients. American J of Hum Genet 45: A179.
- Blin-Wakkach C, Lezot F, Ghoul-Mazgar S, Hotton D, Monteiro S, et al. (2001) Endogenous Msx1 antisense transcript: In vivo and in vitro evidences, structure, and potential involvement in skeleton development in mammals. Pro of the Nat Acad of Sci 98:7336-7341.
- Lidral AC, Reising BC (2002) The role of MSX1 in human tooth agenesis. J Dent Res 81: 274-278.
- Satokata I, Maas R (1994) Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6:348-356.
- Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE (1996) A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 13: 417-421.
- Bergendal B, Klar J, Stecksén-Blicks C, Norderyd J, Dahl N (2011) Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet A 155A: 1616-1622.
- Abid MF, Simpson MA, Petridis C, Cobourne MT, Sharpe PT (2017) Non-syndromic severe hypodontia caused by a novel frameshift insertion mutation in the homeobox of the MSX1 gene. Arch Oral Biol 75: 8-13.
- Mostowska A, Biedziak B, Trzeciak WH (2006) A novel c.581C>T transition localized in a highly conserved homeobox sequence of MSX1: is it responsible for oligodontia? J of Appl Genet 2006; 47:159-164.
- Xuan K, Jin F, Liu YL, Yuan LT, Wen LY, et al. (2008) Identification of a novel missense mutation of MSX1 gene in Chinese family with autosomal-dominant oligodontia. Arch of Oral Bio 53: 773-779.
- Nieminen P, Kotilainen J, Aalto Y, Knuutila S, Pirinen S, et al. (2003) MSX1 gene is deleted in Wolf-Hirschhorn syndrome patients with oligodontia. J Dent Res 82: 1013-1017.
- Van den, Boogaard MJ, Dorland M, Beemer FA, van Amstel HK (2000) MSX1 mutation is associated with orofacialclefting and tooth agenesis in humans. Nat Genet 24:342-343.
- Jumlongras D, Bei M, Stimson JM, Wang WF, DePalma SR, et al. (2001) A nonsense mutation in MSX1 causes Witkop syndrome. Am J Hum Genet 69: 67-74.
- Wong SW, Liu HC, Han D, Chang HG, Zhao HS, et al. (2014) A novel non-stop mutation in MSX1 causing autosomal dominant non-syndromicoligodontia. Mutagenesis 29: 319-323.
- Tatematsu T, Kimura M, Nakashima M, Machida J, Yamaguchi S, et al. (2015) An Aberrant Splice Acceptor Site Due to a Novel Intronic Nucleotide Substitution in MSX1 Gene Is the Cause of Congenital Tooth Agenesis in a Japanese Family. PLoS One 10: e0128227.
- Mostowska A, Biedziak B, Trzeciak WH (2006) A novel mutation in PAX9 causes familial form of molar oligodontia. Eur J Hum Genet 14: 173-179.
- Shahid M, Balto HA, Al-Hammad N, Joshi S, Khalil HS, et al. (2016) Mutations in MSX1, PAX9 and MMP20 genes in Saudi Arabian patients with tooth agenesis. Eur J Med Genet 59: 377-385.
- Kimura M, Machida J, Yamaguchi S, Shibata A, Tatematsu T, et al. (2014) Novel nonsense mutation in MSX1 in familial nonsyndromicoligodontia: subcellular localization and role of homeodomain/MH4. Eur J of Oral Sci 122: 15-20.
- Kamamoto M, Machida J, Yamaguchi S, Kimura M, Ono T, et al. (2011) Clinical and functional data implicate the Arg(151) Ser variant of MSX1 in familial hypodontia. Eur J of Hum Genet19: 844-850.
- Zhang XX, Wong SW, Han D, Feng HL (2015) Simultaneous Occurence of an Autosomal Dominant Inherited MSX1 Mutation and an X-linked Recessive Inherited EDA Mutation in One Chinese Family with Non-syndromicOligodontia. Chin J Dent Res 18:229-234.
- Arte S, Parmanen S, Pirinen S, Alaluusua S, Nieminen P (2013) Candidate gene analysis of tooth agenesis identifies novel mutations in six genes and suggests significant role for WNT and EDA signaling and allele combinations. PLOS ONE 8:e73705.
- Reddy NA, Adusumilli G, Devanna R, Mayur RG, Pichai S, et al. (2013) Msx1 gene variant - its association in isolated hypodontia: A case control genetic study. Ind J of Hum Genet 19:459-464.
- AlFawaz S, Plagnol V, Wong FS, Kelsell DP (2015) A novel frameshift MSX1 mutation in a Saudi family with autosomal dominant premolar and third molar agenesis. Arch of Oral Bio60: 982-988.
- Stapleton P, Weith A, Urbánek P, Kozmik Z, Busslinger M (1993) Chromosomal localization of seven PAX genes and cloning of a novel family member, PAX-9. Nat Genet 3: 292-298.
- Peters H, Neubuser A, Kratochwil K, Balling R (1998) Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes &Dev12: 2735-2747.
- Mastouri MH, De Coster P, Zaghabani A, Trabelsi S, May Y, et al. (2016) Characterization of a novel mutation in PAX9 gene in a family with non-syndromic dental agenesis. Arch Oral Biol 71: 110-116.
- Guala A, Falco V, Breedveld G, De Filippi P, Danesino C (2008) Deletion of PAX9 and oligodontia: a third family and review of the literature. Int J Paediatr Dent 18: 441-445.
- Kapadia H, Mues G, D’Souza R (2007) Genes affecting tooth morphogenesis. Ortho &Cran Res 10: 237-244.
- Das P, Stockton DW, Bauer C, Shaffer LG, D’Souza RN, et al. (2002)Haploinsufficiency of PAX9 is associated with autosomal dominant hypodontia. Hum Genet 110:371-376.
- Klein ML, Nieminen P, Lammi L, Niebuhr E, Kreiborg S (2005) Novel mutation of the initiation codon of PAX9 causes oligodontia. J Dent Res 84: 43-47.
- Wang Y, Wu H, Wu J, Zhao H, Zhang X, et al. (2009) Identification and functional analysis of two novel PAX9 mutations. Cells Tissues Organs 189: 80-87.
- Wang SK, Chan HC, Makovey I, Simmer JP, Hu JC (2012) Novel PAX9 and COL1A2 missense mutations causing tooth agenesis and OI/DGI without skeletal abnormalities. PLoS One 7:e51533.
- Mostowska A, Zadurska M, Rakowska A, Lianeri M, JagodziÅ, Ski PP (2013) Novel PAX9 mutation associated with syndromic tooth agenesis. Eur J Oral Sci 121: 403-411.
- Lammi L, Halonen K, Pirinen S, Thesleff I, Arte S, et al. (2003) A missense mutation in PAX9 in a family with distinct phenotype of oligodontia. Eur J Hum Genet 11: 866-871.
- Liang J, Song G, Li Q, Bian Z (2012) Novel missense mutations in PAX9 causing oligodontia. Arch Oral Biol 57: 784-789.
- Jumlongras D, Lin JY, Seidman CE, Seidman JG, Maas RL, et al. (2004) A novel missense mutation in the paired domain of PAX9 causes nonsyndromicoligodontia. Hum Genet 114:242-249.
- Zhao JL, Chen YX, Bao L, Xia QJ, Wu TJ, et al. (2005) [Novel mutations of PAX9 gene in Chinese patients with oligodontia]. Zhonghua Kou Qiang Yi XueZaZhi 40: 266-270.
- Zhao J, Hu Q, Chen Y, Luo S, Bao L, et al. (2007) A novel missense mutation in the paired domain of human PAX9 causes oligodontia. Am J Med Genet A 143A: 2592-2597.
- Mostowska A, Kobielak A, Biedziak B, Trzeciak WH (2003) Novel mutation in the paired box sequence of PAX9 gene in a sporadic form of oligodontia. Eur J Oral Sci 111: 272-276.
- Tallon-Walton V, Manzanares-Cespedes MC, Arte S, Carvalho-Lobato P, Valdivia-Gandur I,et al. (2007) Identification of a novel mutation in the PAX9 gene in a family affected by oligodontia and other dental anomalies. Eur J of Oral Sci 115:427-432.
- Mostowska A, Biedziak B, Trzeciak WH (2006) A novel mutation in PAX9 causes familial form of molar oligodontia. Eur J Hum Genet 14: 173-179.
- Suda N, Ogawa T, Kojima T, Saito C, Moriyama K (2011) Non-syndromicoligodontia with a novel mutation of PAX9. J Dent Res 90: 382-386.
- Mitsui SN, Yasue A, Masuda K, Watanabe K, Horiuchi S, et al. (2014) Novel PAX9 mutations cause non-syndromic tooth agenesis. J Dent Res 93: 245-249.
- Zhu J, Yang X, Zhang C, Ge L, Zheng S (2012) A novel nonsense mutation in PAX9 is associated with sporadic hypodontia. Mutagene 27:313-317.
- Boeira Junior BR, Echeverrigaray S (2012) Polymorphism in the MSX1 gene in a family with upper lateral incisor agenesis. Arch Oral Biol 57: 1423-1428.
- Pawlowska E, Janik-Papis K, Poplawski T, Blasiak J, Szczepanska J (2010) Mutations in the PAX9 gene in sporadic oligodontia. OrthodCraniofac Res 13: 142-152.
- Frazier-Bowers SA, Guo DC, Cavender A, Xue L, Evans B, et al. (2002) A novel mutation in human PAX9 causes molar oligodontia. J Dent Res 81: 129-133.
- Zhang W, Qu HC, Zhang Y (2014) PAX-9 polymorphism may be a risk factor for hypodontia: a meta-analysis. Genet Mol Res 13: 9997-10006.
- Dong X, Seelan RS, Qian C, Mai M, Liu W (2001) Genomic structure, chromosome mapping and expression analysis of the human AXIN2 gene. Cyto and Cell Genet 93:26-28.
- Callahan N, Modesto A, Meira R, Seymen F, Patir A, et al. (2009) Axis inhibition protein 2 (AXIN2) polymorphisms and tooth agenesis. Arch Oral Biol 54: 45-49.
- Mostowska A, Biedziak B, Jagodzinski PP (2006) Axis inhibition protein 2 (AXIN2) polymorphisms may be a risk factor for selective tooth agenesis. J Hum Genet 51: 262-266.
- Wong S, Liu H, Bai B, Chang H, Zhao H,et al. (2014) Novel missense mutations in the AXIN2 gene associated with non-syndromicoligodontia. Arch of Oral Bio 59:349-353.
- Rivera B, Perea J, Sánchez E, Villapún M, Sánchez-Tomé E, et al. (2014) A novel AXIN2 germline variant associated with attenuated FAP without signs of oligondontia or ectodermal dysplasia. EurJ of Hum Genet 22: 423-426.
- Marvin ML, Mazzoni SM, Herron CM, Edwards S, Gruber SB, et al. (2011) AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J of Med Genet Part A 155A: 898-902.
- Oklü R, Hesketh R (2000) The latent transforming growth factor beta binding protein (LTBP) family. Biochem J 352: 601-610.
- Yin W, Smiley E, Germiller J, Mechan RP, Florer JB, et al. (1995) Isolation of a novel latent transforming growth factor-beta binding protein gene (LTBP-3). J of Bio Chem 270:10147-10160.
- Noor A, Windpassinger C, Vitcu I, Orlic M, Rafiq MA, et al. (2009)Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-beta binding protein 3. Am J of Hum Genet 84:519-523.
- Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, et al. (1996) X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 13: 409-416.
- Mikkola ML, Thesleff I (2003) Ectodysplasin signaling in development. Cytokine Growth Factor Rev 14: 211-224.
- Orange JS, Levy O, Geha RS (2005) Human disease resulting from gene mutations that interfere with appropriate nuclear factor-kappaB activation. Immun Rev 203: 21-27.
- He H, Han D, Feng H, Qu H, Song S, et al. (2013) Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS One 8: e80393.
- Yang Y, Luo L, Xu J, Zhu P, Xue W, et al. (2013) Novel EDA p.Ile260Ser mutation linked to non-syndromichypodontia. J Dent Res 92: 500-506.
- Mues G, Bonds J, Xiang L, Vieira AR, Seymen F, et al. (2014) The WNT10A gene in ectodermal dysplasias and selective tooth agenesis. Am J of Med Genet Part A 164A: 2455-2460.
- Tarpey P, Pemberton TJ, Stockton DW, Das P, Ninis V, et al. (2007) A novel Gln358Glu mutation in Ectodysplasin A associated with X-linked dominant incisor hypodontia. Am J of Med Genet Part A 143:390-394.
- Lee KE, Ko J, Shin TJ, Hyun HK, Lee SH, et al. (2014) Oligodontia and curly hair occur with ectodysplasin-a mutations. J Dent Res 93: 371-375.
- Kowalczyk-Quintas C, Willen L, Dang AT, Sarrasin H, Tardivel A, et al. (2014) Generation and characterization of function-blocking anti-Ectodysplasin A (EDA) monoclonal antibodies that induce Ectodermal Dysplasia. J of Bio Chem289:4273-4285.
- Li S, Li J, Cheng J, Zhou B, Tong X, et al. (2008) Non-syndromic tooth agenesis in two Chinese families associated with novel missense mutations in the TNF domain of EDA (ectodysplasin A). PLoS One 3: e2396.
- Tao R, Jin B, Guo SZ, Qing W, Feng GY, et al. (2006) A novel missense mutation of the EDA gene in a Mongolian family with congenital hypodontia. J Hum Genet 51: 498-502.
- He H, Han D, Feng H, Qu H, Song S, et al. (2013) Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS One 8: e80393.
- Nikopensius T, Annilo T, Jagomägi T, Gilissen C, Kals M, et al. (2013) Non-syndromic tooth agenesis associated with a nonsense mutation in ectodysplasin-A (EDA). J Dent Res 92: 507-511.
- Rasool M, Schuster J, Aslam M, Tariq M, Ahmad I, et al. (2008) A novel missense mutation in the EDA gene associated with X-linked recessive isolated hypodontia. J of Hum Genet 53: 894-898.
- Kieri CF, Bergendal B, Lind LK, Schmitt-Egenolf M, Stecksén-Blicks C (2014) EDAR-induced hypohidrotic ectodermal dysplasia: a clinical study on signs and symptoms in individuals with a heterozygous c.1072C?>?T mutation. BMC Med Genet16: 15-57.
- Cawthorn WP, Bree AJ, Yao Y, Du B, Hemati N, et al. (2012) Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 50: 477-489.
- Liu F, Millar SE (2010) Wnt/beta-catenin signaling in oral tissue development and disease. J Dent Res 89: 318-330.
- Liu F, Chu EY, Watt B, Zhang Y, Gallant NM, et al. (2008) Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. DevBiol 313: 210-224.
- Jarvinen E, Salazar-Ciudad I, Birchmeier W, Taketo MM, Jernvall J, et al. (2006) Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signaling. Proc of the Nat Acad of Sci 103: 18627-18632.
- Yamashiro T, Zheng L, Shitaku Y, Saito M, Tsubakimoto T, et al. (2007) Wnt10a regulates dentin sialophosphoprotein mRNA expression and possibly links odontoblast differentiation and tooth morphogenesis. Differen 75: 452-462.
- Kantaputra P, Sripathomsawat W (2011) WNT10A and isolated hypodontia. Am J Med Genet A 155A: 1119-1122.
- van den Boogaard MJ, Créton M, Bronkhorst Y, van der Hout A, Hennekam E, et al. (2012) Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet 49: 327-331.
- Cluzeau C, Hadj-Rabia S, Jambou M, Mansour S, Guigue P, et al. (2011) Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mutat 32: 70-72.
- Abdalla EM, Mostowska A, JagodziÅ, Ski PP, Dwidar K, Ismail SR (2014) A novel WNT10A mutation causes non-syndromichypodontia in an Egyptian family. Arch Oral Biol 59: 722-728.
- Mu YD, Xu Z, Contreras CI, McDaniel JS, Donly KJ, et al. (2013) Mutational analysis of AXIN2, MSX1, and PAX9 in two Mexican oligodontia families. Genet and Mol Res 12: 4446-4458.
- Satokata I, Maas R (1994) Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6:348-356.
- Nakatomi M, Wang XP, Key D, Lund JJ, Turbe-Doan A, et al. (2010) Genetic interactions between Pax9 and Msx1 regulate lip development and several stages of tooth morphogenesis. Dev Bio 340: 438-449.
- Ogawa T, Kapadia H, Feng JQ, Raghow R, Peters H, et al. (2006) Functional consequences of interactions between Pax9 and Msx1 genes in normal and abnormal tooth development. J of Bio Chem 281: 18363-18369.
- Kapadia H, Frazier–Bowers S, Ogawa T, D’Souza RN (2006) Molecular characterization of a novel PAX9 missense mutation causing posterior tooth agenesis. Eur J of Hum Genet14:403-409.