Keywords
Cysts; Fibromatosis, Aggressive; Pancreas
Abbreviations
DT: desmoid tumor; FAP: familial adenomatous polyposis; PDGFRA: platelet-derived growth factor receptor alpha
INTRODUCTION
Desmoid tumors (DTs) are a rare type of benign soft tissue neoplasia, composed of mature fibroblast proliferation and a dense collagen intercellular matrix [1]. They usually involve fascial and musculo-aponeurotic tissues and belong to the fibromatosis process [2]. Their evolution is characterized by local invasion without metastatic potential [1]. DTs account for 0.03% of all neoplasms and 3% of soft tissue tumors, and their annual incidence has been estimated to be 2-4 cases per million per year in the general population.
Intra-abdominal DTs are most often associated with familial adenomatous polyposis or Gardner syndrome (familial adenomatous polyposis with multiple osteomas and mesenchymal tumors of the skin and soft tissues) in up to 70% of the cases while sporadic cases are uncommon [3, 4, 5]. Multiple reports have suggested a link between DTs and hormonal factors or local traumatism [6, 7]. DTs usually grow asymptomatically for a long period of time before a diagnosis can be made on the basis of symptoms and/or mechanical complications [1]. The management of DTs requires special attention. Indeed, recurrence after surgical resection occurs in up to 85% of cases and it must be considered only for rapidly growing and life-threatening tumors [8, 9]. Tamoxifen, toremifene, and sulindac and cytotoxic chemotherapy and radiation therapy are commonly reported as effective treatment for DTs and are considered to be first line treatments [6].
DTs occur very rarely in the pancreas [10, 11, 12, 13, 14, 15, 16]. We report a case which developed in a 51-year-old woman, presenting with recurrent abdominal pain and weight loss.
CASE REPORT
A 51-year-old woman was referred to our institution after a 2-month period of progressive epigastric pain with dorsal irradiation, nausea, vomiting and weight loss. Her past medical history revealed a conservative hysterectomy in 1993 for a symptomatic benign leiomyofibroma. No family history suggestive of a genetic disease was found.
Clinical examination was normal. Serum lipase and liver tests were normal as were upper gastrointestinal endoscopy and ileocolonoscopy. Spiral computed tomography (CT) revealed a homogeneous, 6 cm mass in the pancreatic tail, mainly solid but with a cystic component (Figure 1). The pancreatic mass was well-delimited but not encapsulated. There was no sign of locoregional invasion or metastasis. The surrounding vessels (superior mesenteric artery, celiac trunk, and splenic and portal veins) were not invaded. Endoscopic ultrasonography showed a hypoechoic, welldemarcated lesion. The surrounding pancreas was normal. The serum tumor markers were normal: serum carcinoembryonic antigen, 1.4 ng/mL (reference range: 0-3 ng/mL); carbohydrate 15-3, 11 UI/mL (reference range: 0-30 UI/mL); and carbohydrate 19-9, 31 UI/mL (reference range: 0-37 UI/mL). The diagnosis of mucinous cystadenocarcinoma was suspected and a distal pancreatectomy was performed.
Figure 1. Spiral computed tomography scans at initial
presentation. Presence of a homogeneous, 6 cm mass
of the pancreatic tail (arrow-head), surrounded by a
cystic component (stars).
Surgical exploration confirmed a 6 cm lesion of the pancreatic tail, adherent to the posterior wall of the stomach, the mesocolon and the duodenojejunal junction. A left splenopancreatectomy with en-bloc resection of the splenic colonic flexure, duodenojejunal junction and part of the posterior gastric wall was performed. The macroscopic examination revealed a poorly circumscribed, nonencapsulated, dense, fasciculated tumor. The transection margin was tumor-free.
Histological analysis showed that the mass had infiltrated the surrounding pancreatic parenchyma (Figure 2ab) and consisted of spindle cells with a regular nucleus, separated by large amounts of collagen fibers in edematous tissue with rare inflammatory cells (Figure 2c). No necrosis was found and mitotic activity was low (less than 2 per 10 high power fields). The cystic component was related to retentional cysts and duct dilatation, within or adjacent to the tumor. The tumor invaded the muscularis propria of both the stomach and the small bowel. All seven lymph nodes were tumor-free. Complete pathological examination revealed no angioor nervous invasion. The pathological features were consistent with a desmoid tumor.
Figure 2. Histological (a and b) and immunohistochemical (c-f) findings of the pancreatic desmoid tumor showing a
mesenchymatous tumor in contact with the normal pancreas (a.) largely dissecting the parenchyma (b.). Highmagnification
showed that the mass consisted of spindle cells, with a regular nucleus, separated by large amounts of
collagen fibers in edematous tissue with rare inflammatory cells (hematoxylin-eosin stain, original magnification x400)
(c.). All tumor cells were negative with monoclonal anti-CD34 antibody (d.). Moderate nuclear staining was detected
with beta-catenin antibody (e.). All tumor cells were negative for Bcl-2 (f.).
Immunohistochemical analysis revealed that the tumor cells did not express anti-CD34 (QBEnd10 clone, Immunotech, Marseille, France) (Figure 2d), anti-S100 protein (polyclonal, Immunotech, Marseille, France), anti-desmin (D33 clone, Dako Co., Kyoto, Japan), anti-alpha smooth muscle actin (1A4 clone, Dako Co., Kyoto, Japan), anti-Bcl2 (124-(3) clone, Dako Co., Kyoto, Japan) (Figure 2e) and anti-caldesmon (h-CD clone, Dako Co., Kyoto, Japan) antibodies. Moderate, cytoplasmic staining was found with anti-CD117 antibody (c-kit, Dako Co., Kyoto, Japan) in 10 to 20% of tumor cells. All tumor cells were positive (nuclear staining) for anti-beta-catenin (C19220 clone, Becton Dickinson Transduction Laboratories, Cowley, UK) (Figure 2f) and were negative for anti-MDM2 (IF2 clone, Zymed Laboratories, San Francisco, CA, USA) and anti-CDK4 antibodies (DCS-31 clone, Biosource International, Camarillo, CA, USA).
Mutational analysis of the alpha chain of the PDGF receptor (PDGFRA) (exons 12, 14 and 18) c-kit (exons 9, 11, 13 and 17) and betacatenin were performed on frozen material. No mutation was found for PDGFRA and ckit genes. A T41A substitution of the betacatenin gene was demonstrated (Figure 3).
Figure 3. Electrophoregram of the beta-catenin gene
sequencing, carried out using a polymerase chain
reaction with the following primers: 5’-
ATGGAACCAGACAGAAAAG-3’, 5’-TACAGGACT
TGGGAGGTATC-3’, 5’-AACCAGACAGAAAGCGG-3
and 5’-GACTTGGGAGGATCCACA-3’. The resulting
sequence data were analyzed and compared to a normal
sequence. In our case, we observed a T41A
substitution.
After surgery, the patient developed a lowoutput asymptomatic pancreatic fistula which was treated conservatively. Oral alimentation was started on day 7 and she was discharged on day 20. A short term follow-up showed rapid disappearance of the symptoms which had revealed the DT. Because of complete resection and the sporadic origin of the DT, no complementary treatment was given. After 18-months of follow-up, clinical examination and CT scan were normal.
DISCUSSION
DT is a rare form of connective tissue tumor, developed from fascial and aponeurotic tissue, also called a fibromatosis process [2]. A DT is a benign neoplasia, characterized by local growth [1]. Whereas DT has no metastatic potential, local invasion can provide mechanical complications such as compression and/or obstruction of blood vessels, or the digestive or urinary tract. Multiple DTs, major surgery and DT recurrences can be life-threatening [3]. DTs can be divided into three different localizations: DT of the abdominal or pelvic wall (49% of cases), extra-abdominal DT (shoulder girdle, chest and inguinal region in 43% of cases) and intra abdominal DT (retroperitoneum and mesentery in 8% of cases) [17].
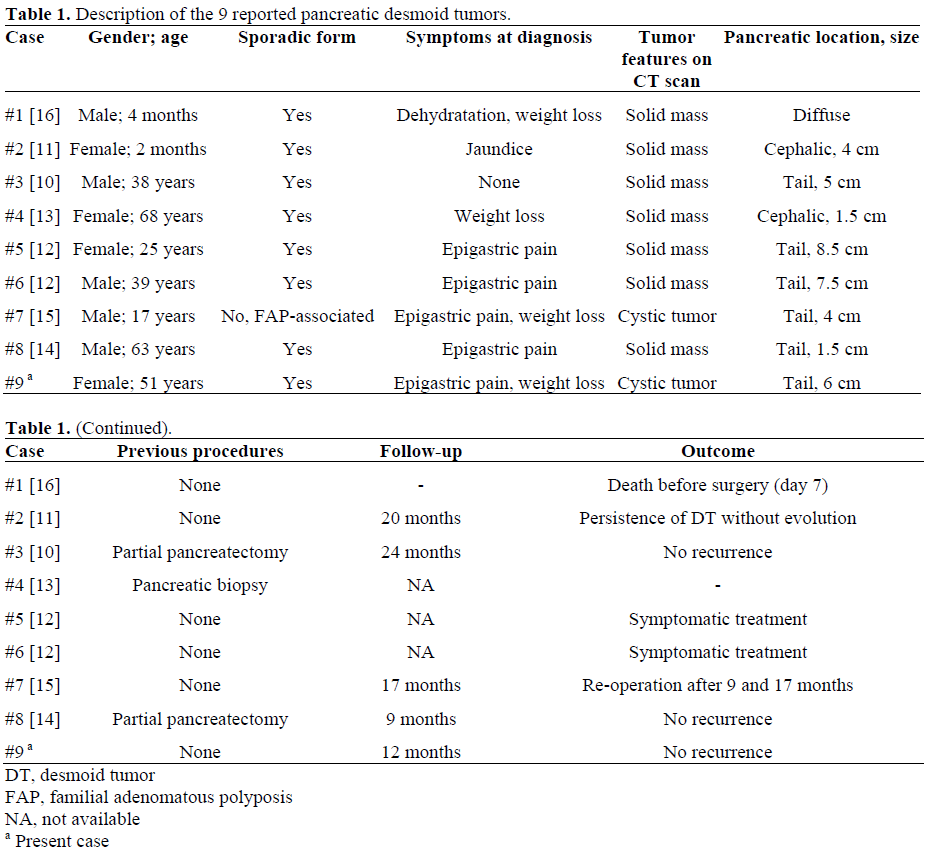
Pancreas is a very rare location for DT. Eight cases have previously been reported involving the pancreas (Table 1). Interestingly, while intra-abdominal DTs are mostly associated with familial adenomatous polyposis, such association was found in only one of the 8 pancreatic DTs [15]. Our case is in line with this finding. Interestingly, pancreatic DTs occurred subsequent to pancreatic surgery or biopsy in three cases [12, 13]. The symptoms were mainly related to epigastric pain (4/8) and weight loss (3/8). The DTs were mostly localized in the tail (5/8) and measured 30-85 mm. A laparotomy was performed in all patients, permitting tumor biopsy (n=3) or pancreatic resection (n=5).
Differentiating DTs from other types of soft tissue neoplasms may be difficult based on histological examination alone (Table 2). Immunohistochemistry and molecular biology are therefore very useful. The negativity of bcl2, CD34, CD117, MD-M2 and CDK4 expression excluded a solitary fibrous tumor, a gastrointestinal stromal tumor (GIST) or a dedifferentiated liposarcoma [18, 19]. In c-kit negative tumors, mutation in c-kit or PDGFRA genes can be assessed by molecular pathology and allow us to distinguish GISTs from other soft tissue tumors. The absence of the mutation of PDGFRA and c-kit genes excluded the diagnosis of a GIST in our case [20, 21]. DT are supposed to be related to clonal myofibroblastic proliferation and to the somatic mutation of the Wnt/beta-catenin gene, leading to the nuclear accumulation of beta-catenin. A recent study demonstrated that nuclear beta-catenin immunostaining is efficient in distinguishing DTs from other benign and malignant fibroblastic and myofibroblastic lesions [22]. In our case, the nuclear immunostaining of beta-catenin and the mutation of the beta-catenin gene were in accordance with these data and supported the diagnosis of a DT.

In this case, the initial diagnosis was a mucinous cystadenocarcinoma. This was suggested because of the cystic component, the tail location and the female gender. In fact, the cystic component corresponded to retentional cysts, located above the tumor. Only two published pancreatic DTs presented as cystic tumors [15]. In the first case, the cystic component corresponded to a benign retentional cyst as in our patient. The other case corresponded to an intraductal papillary mucinous neoplasia adjacent to a DT with no true intratumoral cystic component [13]. These cases confirm the difficulties of diagnosis of incidental cystic pancreatic tumors.
In the case of extensive invasion, potential life-threatening complications or medical treatment failure, surgical resection is generally performed. However, difficulties can occur because of extensive mesentery or retroperitoneal involvement [3]. Recurrence occurs in some patients, depending on margin-free resection, age, location and associated familial adenomatous polyposis [4, 8]. In the case of intra-abdominal DTs, the recurrence rate could reach 90% whereas, in the case of familial adenomatous polyposisassociated and sporadic DTs, it could reach 10% [23]. Treatment of pancreatic DT in the previous series consists of surgical resection in 6/8 cases with no significant perioperative complications. Resection was carried-out in 5 patients and consisted in partial pancreatectomy except for one case of total pancreatectomy. The remaining patient did not present post-operative DT growth. The recurrence occurred in the only patient presenting a past history of familial adenomatous polyposis. However, the postoperative follow-up was limited [11, 12, 13, 14, 15, 16]. This is consistent with the recurrence rate of intra-abdominal DTs not localized in the pancreas [3, 4]. In the current case, a sporadic form of DT, there was a one year follow-up after surgery with no evidence of recurrence.
In conclusion, DTs are very rare in the pancreas and their diagnosis can be difficult such as in our case where it presented as a cystic lesion. In contrast to intra-abdominal forms, sporadic pancreatic DTs are more frequent than those associated with familial adenomatous polyposis.
Conflict of interest
The authors have no potential conflicts of interest
References
- Clark SK, Phillips RK. Desmoids in familial adenomatous polyposis. Br J Surg 1996; 83:1494-504. [PMID: 9014661]
- Goellner JR, Soule EH. Desmoid tumors, an ultrastructural study of eight cases. Hum Pathol 1980; 11:43-50. [PMID: 7364438]
- Clark SK, Neale KF, Landgrebe JC, Phillips RK. Desmoid tumours complicating familial adenomatous polyposis. Br J Surg 1999; 86:1185-9. [PMID: 10504375]
- Nieuwenhuis MH, De Vos Tot Nederveen Cappel W, Botma A, Nagengast FM, Kleibeuker JH, Mathus- Vliegen EM, et al. Desmoid tumors in a dutch cohort of patients with familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:215-9. [PMID: 18237870]
- Hartley JE, Church JM, Gupta S, McGannon E, Fazio VW. Significance of incidental desmoids identified during surgery for familial adenomatous polyposis. Dis Colon Rectum 2004; 47:334-8. [PMID: 14991495]
- Rampone B, Pedrazzani C, Marrelli D, Pinto E, Roviello F. Updates on abdominal desmoid tumors. World J Gastroenterol 2007; 13:5985-8. [PMID: 18023087]
- Cohen S, Ad-El D, Benjaminov O, Gutman H. Post-traumatic soft tissue tumors: case report and review of the literature a propos a post-traumatic paraspinal desmoid tumor. World J Surg Oncol 2008; 6:28. [PMID: 18312655]
- Ballo MT, Zagars GK, Pollack A, Pisters PW, Pollack RA. Desmoid tumor: prognostic factors and outcome after surgery, radiation therapy, or combined surgery and radiation therapy. J Clin Oncol 1999; 17:158-67. [PMID: 10458229]
- Kulaylat MN, Karakousis CP, Keaney CM, McCorvey D, Bem J, Ambrus Sr JL. Desmoid tumour: a pleomorphic lesion. Eur J Surg Oncol 1999; 25:487- 97. [PMID: 10527597]
- Bruce JM, Bradley EL 3rd, Satchidanand SK. A desmoid tumor of the pancreas. Sporadic intraabdominal desmoids revisited. Int J Pancreatol 1996; 19:197-203. [PMID: 8807365]
- Ure BM, Holschneider AM, Gharib M, Halsband H, Hinselmann D. Clinical aspects, classification and prognosis of 7 cases of pediatric fibromatosis. Z Kinderchir 1988; 43:27-30. [PMID: 3376585]
- Nursal TZ, Abbasoglu O. Sporadic hereditary pancreatic desmoid tumor: a new entity? J Clin Gastroenterol 2003; 37:186-8. [PMID: 12869894]
- Sedivy R, Ba-Ssalamah A, Gnant M, Hammer J, Klöppel G. Intraductal papillary-mucinous adenoma associated with unusual focal fibromatosis: a "postoperative" stromal nodule. Virchows Arch 2002; 441:308-11. [PMID: 12242530]
- Weiss ES, Burkart AL, Yeo CJ. Fibromatosis of the remnant pancreas after pylorus-preserving pancreaticoduodenectomy. J Gastrointest Surg 2006; 10:679-88. [PMID: 16773761]
- Pho LN, Coffin CM, Burt RW. Abdominal desmoid in familial adenomatous polyposis presenting as a pancreatic cystic lesion. Fam Cancer 2005; 4:135- 8. [PMID: 15951964]
- Roggli VL, Kim HS, Hawkins E. Congenital generalized fibromatosis with visceral involvement. A case report. Cancer 1980; 45:954-60. [PMID: 7260846]
- Weiss S, Goldblum JR. Liposarcoma. In: Enzinger and Weiss’s Soft Tissue Tumors, 4th ed. St. Louis: Mosby, 2001: 641-93. [ISBN: 0323012000]
- Shimada S, Ishizawa T, Ishizawa K, Matsumura T, Hasegawa T, Hirose T. The value of MDM2 and CDK4 amplification levels using real-time polymerase chain reaction for the differential diagnosis of liposarcomas and their histologic mimickers. Hum Pathol 2006; 37:1123-9. [PMID: 16938516]
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006; 130:1466-78. [PMID: 17090188]
- Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn Pathol 2006; 23:91-102. [PMID: 17193822]
- Lucas DR, al-Abbadi M, Tabaczka P, Hamre MR, Weaver DW, Mott MJ. c-Kit expression in desmoid fibromatosis. Comparative immunohistochemical evaluation of two commercial antibodies. Am J Clin Pathol 2003; 119:339-45. [PMID: 12645334]
- Bhattacharya B, Dilworth HP, Iacobuzio-Donahue C, Ricci F, Weber K, Furlong MA, et al. Nuclear betacatenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol 2005; 29:653-9. [PMID: 15832090]
- Reitamo JJ. The desmoid tumor. IV. Choice of treatment, results, and complications. Arch Surg 1983; 118:1318-22. [PMID: 6639341]




