Keywords
Epigenetics; Handgrip; Strength; Diabetes mellitus type 2; Cardiovascular disease; Muscle mass
Introduction
In the past decade several large international epidemiological studies were performed in order to determine which risk factors are associated with the presence of cardio-vascular diseases (CVD). The INTERHEART study a case- control study of acute myocardial infarction (AMI), which assessed 52 countries representing every inhabited continent, had a total enrollment of 12,461 cases and 14,637 controls. It showed that smoking, history of hypertension (HTA), diabetes, increased waist/hip ratio, dietary patterns, low physical activity, consumption of alcohol, alterations in the ratio of blood apolipoproteins (APO) B/A1, and psychosocial factors account for more than 90% of the population attributable risk (PAR) for acute myocardial infarction (AMI). These nine modifiable risk factors are similar worldwide, suggesting that most of the cases of AMI are preventable [1]. The INTERSTROKE study was a standardized case- control study in 22 countries; cases were patients with acute first stroke (within 5 days of symptoms onset and 72 h of hospital admission), and controls had no history of stroke, matching them with cases for age and sex. This study revealed that the significant risk factors for all class of strokes are the same as that the observed for AMI and also that together these risk factors account for more than 90% of the PAR for an ischemic or hemorrhagic stroke [2]. Both of these studies concluded that targeted interventions that reduce these risk factor, could substantially reduce both the burden of stroke and CVD.
In recent decades an increased prevalence of CVD mortality has been reported in low-medium income countries, which has been associated with changes in life styles, deficiencies in health systems and the persistence of social inequities. Moreover, recent studies have shown that reduced muscle strength, as measured by hand grip dynamometry (HG) is associated with an increased risk of CVD in diabetes mellitus type 2 (T2DM) patients [3], with high metabolic risk in children [4], and with mortality of all cause and CVD mortality in adults of different regions on the world [5]. Significant differences in HG were observed between people from high, medium and low-income countries, the lowest HG noted in the lowest and highest HG in the highest income countries [5].
We therefore discuss how early life undernutrition, via epigenetic mechanisms has a role in the development of cardiovascular diseases. We also outline the important predictive value of inadequate muscle mass and strength in cardiometabolic disease risk, and suggest that this may mediate associations between poor early conditions and later disease.
Maternal Nutrition as a Regulator of Epigenetic Mechanisms Implicated in CVD
One of the explanations for the increased risk of CVD in lowmedium income countries is maternal under nutrition that promotes fetal intrauterine growth retardation and low birth weight (LBW), which in turn increases the risk of CVD in later life [6,7]; known as The Developmental Origins of Health and Disease (DOHaD) hypothesis [8] (Figure 1). This hypothesis proposes that in response to maternal under nutrition the fetus makes predictive adaptations and develops conservative “intrauterine programming”, prioritizing the development of the central nervous system, potentially at the expense of development of other tissues such as the pancreatic beta cells, nephrons and muscle tissue, which manifests as LBW [9]. However, if the conditions of post uterine life involves exposure to high-energy intakes, the prenatal cues are rendered inappropriate prenatal predictions and postnatal reality are mismatched [8]. In this scenario, the intrauterine metabolic adaptations, or “predictive adaptive responses”, are disadvantageous and instead increase susceptibility to chronic non- communicable disease [10].
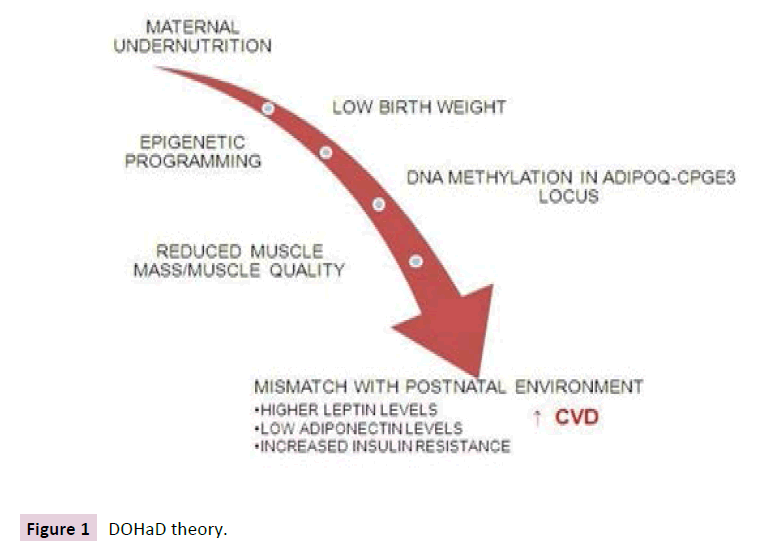
Figure 1 DOHaD theory.
Epigenetics changes are defined as alterations in gene expression without changing the DNA sequence and are the primary basis for developmental programming and the explanatory mechanism for biological changes triggered by socio-economic and environmental conditions [11]. Epigenetics provides a molecular link between prenatal environments, genes, cellular processes and subsequent susceptibility to chronic diseases [12]. Several studies suggest that the epigenetic adaptations may be also permanently passed trans-generationally from mother to progeny [13].
Epigenetics modifications are controlled by changes in DNA methylation and/or chromatin structure, some of which are thought to play an important role in the pathogenesis of various diseases, including cancer and CVD [14]. Epigenetic changes are heritable, although occasionally reversible, depending on endogenous, and especially exogenous (environmental) signals, creating a memory of cell identity, and maintaining genomic functions after differentiation, propagation of essential features of chromosomal architecture, and dosage compensation [15]. Therefore, epigenetic modifications could lead to irreversible processes of differentiation and organogenesis or to labile and potentially reversible changes in homeostatic processes. Moreover, environmental signals such as food depletion and stress have been present throughout evolution, and organisms have had to sense and adapt to them, to ensure their survival. These epigenetic modifications have been implicated in the control of various parameters of gene expression, genetic recombination, DNA repair, and DNA mutagenesis [10]. Epigenetic mechanisms include DNA methylations, histone modifications, and microRNAs [9], and can help to explain how individuals with identical or similar DNA, when exposed to different environmental signals, express diverse phenotypes and differ in their susceptibility to certain pathologies. Thus, chromatin structure can be linked to dietary factors such as nutrient or drug intake, or socio-economic conditions in several ways [6].
Epigenetics and Fetal Programming
Intrauterine programming, regulated by epigenetic mechanisms, is aimed to permit the surviving of the fetus in a medium of maternal under nutrition which may result in intrauterine growth retardation, impaired growth of cells and organs, such as pancreatic beta cells, cardiomiocytes and nephrons, and LBW [9].
Additionally, it has been shown that LBW is related to impaired development of muscle fibers [16], supported by epidemiological studies showing that LBW is associated with lower muscle mass and muscle strength across the lifecycle [17]. Low muscle strength is associated with a poorer cardiometabolic health profile across the lifespan, and with a higher risk of incident disease and mortality in adulthood [4,14].
The next exposure to high energy density food in the postnatal environment may result in rapid catch-up growth associated with an excessive accumulation of ectopic fat, conferring LBW individuals another risk for CVD, T2DM, insulin resistance (IR), HTA and dyslipidemia [4].
DNA modification due to DNA methylation is one of the most widely studied and understood epigenetic mechanisms [18]. It primarily occurs on the cytosine upstream of a guanine (dinucleotide CpG) and is catalyzed by the DNA methyltransferases (DNMTs) [19]. The methylation of cytosines in a CpG context is influenced by both intrauterine stimuli [19,20] and postnatal environmental conditions [21]. Using monozygotic twins, that as definition share genetics and uterus and therefore provides a remarkable investigative example to correlate early life CVD factors and adult life health, Tan et al [22] showed that genome- wide DNA methylation profiling did not reveal epigenetic signatures of birth weight discordance although some sites displayed agedependent intra-pair differential methylation in the extremely discordant twin pairs. It is important to mention that this study is limited by a relatively small sample size for genome- wide association analysis and low coverage of the methylation chips. Future studies that include larger populations and samples from biologically appropriate tissues using next generation sequencing techniques should be able to provide more evidence around the impact of birth weight on DNA methylation in later life and to verify the epigenetic basis of DOHaD [22].
It is important to mention the crucial role of leptin (LEP) and adiponectin (ADIPOQ) genes in metabolism and chronic disease. These genes are the responsible for the encoding of LEP and ADIPOQ proteins, which are produced, stored and released by the adipocytes. LEP’s function is to decreases appetite via hypothalamic receptors. Since LEP levels are increased in obese subjects, it has been proposed that LEP resistance may play a role in the development of obesity and metabolic syndrome [9]. ADIPOQ has insulin-sensitizing effects and anti-inflammatory actions. Low plasma ADIPOQ levels are associated with obesity [23,24], T2DM [25], dyslipidemia [26], increased blood pressure levels [27]. Moreover, it is well documented that ADIPOQ levels are highly heritable (30–70%) [28,29].
Many reports have documented the metabolic effects of variations in the gene coding for adiponectin [30,31]. Metaanalyses of these studies have demonstrated that variability in this gene contributes to the modulation of circulating adiponectin levels and the risk of IR and CVD [28]. However, it is still unclear if these changes are due to genetic inheritance, modifying environmental factors, or simply statistical undulations [28]. Large and collaborative studies are needed to improve understanding.
LEP also acts as a helping neutrophil factor in the central nervous system [32] and it has been proposed as a mayor mediator in the risk of obesity in response to adverse intrauterine stress. In maternal malnutrition, circulating leptin levels are reduced, leading to lower leptin sensitivity and increased vulnerability to obesity in the offspring [33-35]. In contrast, ADIPOQ maintains insulin sensitivity, and its levels are inversely correlated to obesity [36].
Prenatal stress and the exposure to excess glucocorticoids also influence fetal programming and produce changes in the hypothalamic-pituitary-adrenal axis (HPAA) [37]. Prenatal stress due to restricted maternal undernutrition produces intrauterine growth retardation and LBW is associated with increased cortisol levels in fetal blood.
Moreover, LBW is associated with hyperactivity of HPAA during adulthood, leading to metabolic alterations that may promote the subsequent development of T2DM and CVD [38]. It also has been proved that postnatal stress in early-life affects physical health in adulthood including increased risk for obesity, metabolic syndrome, T2DM, CVD, and premature mortality [39,40] via alterations in the HPAA axis, immune system, energy/metabolic regulation [41,42], epigenetic changes [43], and by shortening of telomere length [44]. Inflammatory markers such as C-reactive protein (CRP) and IL-6 are also elevated in young individuals with early-life adversities [45]. Others factors such as cold weather and emotional stress have also been associated with changes in the methylation/acetylation of key promoter genes that regulate the expression of LEP, ADIPOQ, as well as angiotensin II [46].
Insulin-like growth factor II (IGF-II) is a key factor in human development and is maternally imprinted [47]. In conditions of maternal undernutrition there is hypomethylation of the IGF-II loci, due to a decrease of intrauterine supply of methionine or folic acid, which are capable of donating a methyl group (CH3) [19,48], leading to bi-allelic expression of IGF-II, which is associated with an increased risk of colorectal adenoma excessive growth (Beckwith-Wiedemann syndrome) [32]. Recent observation shows that periconceptional administration of folic acid to the mother is related to an increased methylation of the IGF-II gene of the offspring [49]. However, it remains to be determined whether the changes in IGF-II gene methylation are associated with changes in gene expression and birth weight [50]. In contrast, other imprinted genes such as Cyclin-dependent kinase inhibitor 1C (CDKN1C), Pleckstrin homology-like domain family A member 2 (PHLDA2) and Potassium voltage-gated channel (KCNQ1) tend to restrict fetal body´s weight and are associated with restriction growth syndromes such as Silver-Russell syndrome [50]. All these genes are located in the same cluster on chromosome 11 at p15.5, and their regulation its is due to two independent imprinting centers; Imprinting Center 1 and Imprinting Center 2. IGF-II and the imprinted maternally expressed non coding transcript (H19) are localized in the Imprinting Center 1, which contains a methylation- sensitive chromatin insulator that is necessary for controlling the expression of both genes [51]. Dysregulation of the IGF2/H19 imprinted region is related to Beckwith- Wiedemann and Silver-Russell syndrome [51]. Genomic DNA methylation in these imprinting centers may exhibit significant fluctuation among human individuals [52]. It has been shown that IGF- II gene levels were down regulated in Intrauterine Growth Restriction (IUGR), both in fetal and placental samples, which could explain part of the pathology of IUGR [53], although much more investigation is needed to clarify the function of these imprinted genes, their epigenetics and impact in fetal growth and postnatal development.
The Renin Angiotensin System (RAS) is more active in the neonatal period and plays an important role in fetal growth and development [54]. RAS hyperactivity is related to HTN and CVD [55]. Previous studies show that LBW is associated with the modifications of some elements of the RAS. For example, it has been established that Ang II plasma levels in early postnatal life were higher in LBW than in NBW infants predisposing to CVD [56]. The angiotensin-converting enzyme (ACE) level is highly variable between individuals [57] and It has been shown that this variability is associated with an insertion (I)/ deletion (D) polymorphism situated in intron 16 of the ACE gene (ACE/ID polymorphism) [56]. The D allele and the DD genotype are associated with elevated levels of ACE and a higher risk of CVD and end organ damage [55]. It has been established that LBW children had a higher D allele frequency than normal birth weight children (NBW) [58]. LBW children also have higher blood pressure and increased ACE activity levels in comparison with NBW children [57]. Moreover, LBW children have lower levels of DNA methylation in 3 CpG sites from the ACE gene promoter compared with NBW children and the hypomethylation occurs at the same time with an increase of the ACE protein activity and high blood pressure in LBW [57]. Likewise, it has been demonstrated that impairment of growth during prenatal and early postnatal life has been associated with increased plasma concentrations of fibrinogen and factor VII in adulthood [59]. The increased concentrations of these two hemostatic factors found in the plasma may explain why LBW individuals are predisposed to thrombosis and an increased risk of CVD in adulthood [60].
These observations provide solid support for the idea that nutritional deficiencies during critical periods of ontogenic fetal development may have a lifelong influence on health through the regulation in the expression of several genes that result in increased or decreased synthesis of proteins such as angiotensin II (Ang II), LEP, and ADIPOQ [61,62]. As mentioned, the mismatch between a restricted access to nutrients in prenatal life and a calorific excess in postnatal life environment predisposes to rapid catch up growth and a selective accumulation of ectopic fat. Studies have shown that ectopic fat produces an increased level of Ang II that contributes to insulin resistance and water retention, risk factors for HTN and CVD [9,30]. Additionally, Ang II inhibits the differentiation of adipocyte precursors, thus decreasing the percentage of small insulin-sensitive adipocytes and promoting the presence of large adipocytes resistant to the action of insulin and with less potential to produce adiponectin which play critical roles in energy, lipids and glucose homeostasis [32,63].
Muscle Strength and Cardio metabolic Disease
It is well recognized that skeletal muscle is important in the regulation of the glucose and lipid metabolism [7]. Recent findings suggest that muscle may also have a role in the regulation of low degree inflammation through the production of various antiinflammatory myokines [11]. Therefore, muscle tissue counteracts adipocyte as an endocrine organ, by opposing the effects of its adipokines which promote insulin resistance and systemic low degree inflammation, both key processes in the development of atherosclerosis, T2DM and CVD [11]. Indeed, it is suggested that muscle contraction stimulated myokine secretion, is key in mediating the protective effects of physical activity against metabolic risk factors [11]. Myokines are therefore important contributors of the beneficial effects of exercise and are central to the concept of the cross-talk between skeletal muscles and other organs during and after exercise [64]. Therefore, both muscle and fat tissue are described as endocrine organs, which can interact with each other and with the liver, brain, and other organs in the regulation of the glucose and lipid metabolism and pro-antiinflammatory cytokine balance [65].
Low muscle strength evaluated by HG has been shown to be related to higher cardiometabolic risk factors in children from a number of populations [4,66,67], and to be associated with a higher risk of cardiovascular events and mortality in patients with T2DM [4]. Moreover, a recent study that included around 130.000 subjects of 17 high, medium and low-income countries [5] have shown that HG predicted total and cardiovascular mortality. This study also showed race/ethnic differences in HG strength, which also depended on economic classification of the countries, with the lowest HG values observed in non-Caucasian individuals and in low-income countries [5]. Low HG strength is also associated with important non CVD health-related outcomes, such as impaired skeletal health, reduced quality of life and poor mental health [68]. Likewise, in Colombian youth low HG was also inversely associated with the concentrations of triglycerides, C- reactive protein, DBP, HOMA index and metabolic risk score, giving us a potential tool for future primary assessment campaigns to identify individuals at risk and implement primordial preventative programs to reduce T2DM and CVD incidence [5], particularly in LBW individuals [69].
Epigenetics and Skeletal Muscle
Epigenetics has a role in the conformation and expression of several genes in a number of tissues, including muscle. Mature skeletal muscle is a highly plastic tissue that responds to a variety of environmental and nutritional influences [24]. Likewise, maternal undernutrition that subsequently causes LBW and reduced lean mass including lower muscle mass [70]. Epidemiological studies consistently show that LBW is associated with deficits in muscular fitness across the lifecycle [71] including lower muscle strength [72,73], sprint performance [36] and power.
It is clear that LBW is not only associated with smaller muscle tissue mass due to reduced fiber number [13,24,26] but also has other repercussions for muscle, including alterations in muscle fiber type, enzyme content and signaling pathways [74]. Muscle fiber number and type are set in-utero and only the relative proportions of IIa and IIx consistently show post-natal inter-conversion [75], induced fetal under-nutrition in animals is generally associated with lower muscle fiber number, a lower % of type I and higher % of type IIx/b fibers as well as lower muscle mass, and higher fiber size, particularly following rapid post natal catch-up growth [29]. It is important to note however that a larger fiber size may in this scenario reflect higher intra-myocellular lipid content, reported following fetal under-nutrition in animals [76] and in obese adults, [55] rather than the additional myofibrillar protein. A higher proportion of the more insulin resistant and less oxidative type IIx/b fibers, and a lower proportion of the more IIa fibers were reported in healthy, normal-weight young LBW males [77]. Indeed, a lower proportion of type IIa fibres is also associated with obesity in adults [78], and is a characteristic of diabetic rats.
It is evident that skeletal muscle phenotype is affected by the epigenetic changes related to maternal under nutrition, particularly at critical periods of gestation, and it appears that these differences may contribute to an increased susceptibility to metabolic abnormalities and obesity [29]. Nutrients and growth factors are important determinants of muscle mass in adulthood as well as during the early development of skeletal muscle. Evidence from human studies comparing low versus normal birth weight adults and from animal studies of nutrient restriction during gestation show that the epigenetic influences of maternal nutrition impact a number of important metabolic loci in muscle [79]. The mechanistic target of rapamycin (mTOR) signaling pathway for example, is a key loci which coordinates and regulates skeletal muscle protein synthesis [80].
There are two mTOR multiprotein complexes mTORC1 and mTORC2. mTORC1 activation induces cell growth by promoting protein, lipids and nucleotides synthesis, ribosomal biogenesis and inhibiting autophagy. In contrast, mTORC2 controls actin polymerization and the activation of Protein kinase B (Akt) by phospholyrating their hydrophobic motif giving it a catalytic function [79]. Nutrient restriction inhibits mTOR signaling, with consequent decreases in anabolism and increased catabolic processes [25]. The 5' AMP-activated protein kinase enzyme (AMPK) plays a key role in this response and in maintaining metabolic homeostasis, acting as energy sensor sensitive to an increase in ADP/ATP and AMP/ATP ratios associated with nutrient restriction and depletion of intracellular ATP [25]. Under these conditions, AMPK activates catabolic pathways to restore ATP levels [81] and inhibits muscle protein synthesis both via inhibition of the mTOR signaling pathway and by inhibiting elongation factor 2 (eEF2), which is involved in peptide chain elongation in skeletal muscle [24]. Akt is another important signaling pathway that influences muscle protein synthesis by regulating mTOR activity. [82]. LBW rat pups showed a significant reduction in mTOR [83], while impaired Akt signaling was observed in muscle of LBW healthy young adults [84]. Since Akt regulates muscle protein synthesis, its inhibition is a possible mechanism for lifelong deficits in muscle mass observed in LBW individuals. It is important to note that Akt signaling is also an important pathway for insulin action – deficits in which may be a precursor to later insulin resistance.
Conclusion
The results of the studies included in this review suggest that not only the imbalance of the muscle activity as a result of low physical activity, sedentary and western model lifestyle adoption, contributes to the pathogenesis of insulin resistance (IR) and therefore to T2DM and CVD, but also that epigenetics adaptations due to poor intra uterine conditions plays a role in the pathogenesis of T2DM and CVD. In addition to the declining physical activity levels and increased consumption of high-energy- dense food, the populations of developing countries may be more vulnerable to CVD due to these early life epigenetic adaptations.
The reviewed evidence also shows that maternal under nutrition has a crucial impact on muscle mass and muscle quality [74,85], and that epigenetic mechanisms are clearly implicated in the interrelationship between low birth weight, deficits in muscle development and subsequent risk of CVD and other chronic diseases [65].
What still remains unresolved is whether the association between cardiometabolic risk/mortality and HG or other measures of muscle strength is due to the relationship between birth weight and strength (i.e. strength as a marker of poor early conditions) or that strength/muscle mass are key mediators of the well described association between LBW and adult disease (i.e. strength as a direct determinant of health).
References
- Yusuf S, Hawken S, Ôunpuu S, Dans T, Avezum T, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004 Sep 11-17; 364 (9438):937-52.
- O'Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, et al. Risk factors for ischemic and intracerebral hemorrhagic stroke in 22 countries (the INTERSTROKE study), a case-control study. Lancet. 2010 Jul 10; 376 (9735): 112-23.
- López-Jaramillo P, Cohen DD, Gómez-Arbeláez D, Bosch J, Dyal L, Yusuf S, et al. Association of Handgrip strength to cardiovascular mortality in pre-diabetic and diabetic patients: a subanalysis of the ORIGIN trial. Int J Cardiol. 2014 Jun 15; 174(2):458-61.
- Gómez-Arbeláez D, Camacho PA, Cohen DD, Rincón-Romero K, Alvarado- Jurado L, Pinzón S, et al. Higher household income and the availability of electronic devices and transport at home are associated with higher waist circumference in Colombian children: the ACFIES study. Int J Environ Res Public Health. 2014; 11:1834–43.
- Leong DP, Teo KK, Rangarajan S, Lopez-Jaramillo P, Avezum A Jr, Orlandini A, et al. Prognostic value of grip strength: findings from the Prospective Urban Rural Epidemiology (PURE) study. Lancet. 2015 Jul 18; 386(9990):266-73.
- Lopez-Jaramillo P, Gomez-Arbelaez D, Sotomayor-Rubio A, Mantilla-Garcia D, Lopez-Lopez J. Maternal undernutrition and cardiometabolic disease: A latin american perspective. BMC Medicine. 2015;13(1):41.
- Benyshek DC. The “Early Life” Origins of Obesity-Related Health Disorders: New Discoveries Regarding the intergenerational Transmission of Developmentally Programmed Traits in the Global Cardiometabolic Health Crisis. Am J Phys Anthropol. 2013 Dec; 152 Suppl 57:79-93.
- Gluckman PD, Hanson MA, Beedle AS, Spencer HG. Predictive adaptive responses in perspective. Trends Endocrinol Metab. 2008 May-Jun;19(4):109-10.
- Lopez-Jaramillo P, Lahera V, Lopez-Lopez J. Epidemic of cardiometabolic diseases: a Latin American point of view. Ther Adv Cardiovasc Dis. 2011; 5:119–31
- Vickers M. Early Life Nutrition, Epigenetics and Programming of Later Life Disease. Nutrients.2014 Jun 2; 6(6):2165-78.
- Eckardt K, Görgens S, Raschke S and Eckel J. Myokines in insulin resistance and type 2 diabetes. Diabetologia. 2014 Jun; 57(6):1087-99.
- Cianfarani S, Agostoni C, Bedogni, Canani R, Brambilla P, Nobili V, et al. Effect of intrauterine growth retardation on liver and long-term metabolic risk. Int J Obes (Lond).2012 Oct; 36(10):1270-7.
- Bygren LO, Kaati G, Edvinsson S. Longevity determined by paternal ancestors' nutrition during their slow growth period. Acta Biotheor. 2001 Mar;49(1):53-9.
- Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009 Oct; 9(10):692-703.
- Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2. Reproductive Toxicology 2008 Jan; 25(1):2-6.
- Brameld JM. The influence of undernutrition on skeletal muscle development. Br J Nutr 2004; 91: 327–8.
- Gale CR, Martyn CN, Cooper C, Sayer AA. Grip strength, body composition, and mortality. Int J Epidemiol 2007; 36: 228–35.
- Houde AA,Légaré C, Biron S, Lescelleur O, Biertho L, Marceau S,et al. Leptin and adiponectin DNA methylation levels in adipose tissues and blood cells are associated with BMI,waistgirth and LDL-cholesterol levels in severely obese men and women. BMC Med Genet. 2015 May 1; 16:29.
- Tobi EW1, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009 Nov 1;18(21):4046-53.
- Pilsner JR1, Hall MN, Liu X, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Yunus M, Rahman M, Graziano JH, Gamble MV. Influence of prenatal arsenic exposure and newborn sex on global methylation of cord blood DNA. PLoS One. 2012;7(5):e37147. doi: 10.1371/journal.pone.0037147. Epub 2012 May 25.
- Gillberg L , Jacobsen SC ,Rönn T , Brøns C , Vaag A. PPARGC1A DNA methylation in subcutaneous adipose tissue in low birth weight subjects--impact of 5 days of high-fat overfeeding. Metabolism.2014 Feb 63(2):263-71.
- Tan Q, Frost M, Heijmans B, Hjelmborg J, Tobi E, Christensen K and Christiansen L. Epigenetic signature of birth weight discordance in adult twins. BMC Genomics 2014, 15:1062.
- Yang W-S, Lee W-J, Funahashi T, Tanaka S, Matsuzawa Y, Chao C-L, et al. Plasma Adiponectin levels in overweight and obese Asians. Obesity Research. Wiley-Blackwell; 2002 Nov;10(11):1104–10
- Koh S-B, Yoon J, Kim J-Y, Yoo B-S, Lee S-H, Park J-K, et al. Relationships between serum Adiponectin with metabolic syndrome and components of metabolic syndrome in non-diabetic Koreans: ARIRANG study. Yonsei Medical Journal. Yonsei University College of Medicine (KAMJE); 2011;52(2):234.
- Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, Adipose-Specific protein, Adiponectin, in type 2 diabetic patients. Arteriosclerosis, Thrombosis, and Vascular Biology. Ovid Technologies (Wolters Kluwer Health); 2000 Jun 1;20(6):1595–9.
- 26. Matsubara M, Maruoka S, Katayose S. Decreased plasma Adiponectin concentrations in women with Dyslipidemia. The Journal of Clinical Endocrinology & Metabolism. The Endocrine Society; 2002 Jun;87(6):2764–9
- Kazumi T, Kawaguchi A, Sakai K, Hirano T, Yoshino G. Young men with high- normal blood pressure have lower serum Adiponectin, smaller LDL size, and higher elevated heart rate than those with optimal blood pressure. Diabetes Care. American Diabetes Association; 2002 Jun 1;25(6):971–6.
- Comuzzie AG, Funahashi T, Sonnenberg G, Martin LJ, Jacob HJ, Black AEK, et al. The genetic basis of plasma variation in Adiponectin, a global Endophenotype for obesity and the metabolic syndrome. The Journal of Clinical Endocrinology & Metabolism. The Endocrine Society; 2001 Sep;86(9):4321–5.
- Menzaghi C, Trischitta V, Doria A. Genetic influences of Adiponectin on insulin resistance, type 2 diabetes, and cardiovascular disease. Diabetes. American Diabetes Association; 2007 Feb 15;56(5):1198–209.
- Tso A, Sham P, Wat N, Xu A, Cheung B, Rong R, et al. Polymorphisms of the gene encoding adiponectin and glycaemic outcome of Chinese subjects with impaired glucose tolerance: A 5-year follow-up study. Diabetologia 2006. Jun 22.
- Sutton B, Weinert S, Langefeld C, Williams A, Campbell J, Saad M, et al. Genetic analysis of adiponectin and obesity in Hispanic families: The IRAS family study. Human genetics. 2005 Apr 22;(117).
- Briffa JF, McAinch AJ, Romano T, Wlodek ME, Hryciw DH. Leptin in pregnancy and development: a contributor to adulthood disease? Am J Physiol Endocrinol Metab. 2015; 308(5):E335–E350.
- Wild SH, Byrne CD. Evidence for fetal programming of obesity with a focus on putative mechanisms. Nutr Res Rev. 2004; 17(2):153–162.
- Barbero A, Astiz S, Lopez-Bote CJ, et al. Maternal malnutrition and offspring sex determine juvenile obesity and metabolic disorders in a swine model of leptin resistance. PloS One. 2013;8(10):e78424.
- Shasa DR, Odhiambo JF, Long NM, Tuersunjiang N, Nathanielsz PW, Ford SP. Multigenerational impact of maternal overnutrition/obesity in the sheep on the neonatal leptin surge in granddaughters. Int J Obes (Lond). 2015;39(4):695–701.
- 36. Hou M, Chu Z, Liu T, et al. A high-fat maternal diet decreases adiponectin receptor-1 expression in offspring. J Matern Fetal Neonatal Med. 2015;28(2):216–221.
- Seckl JR, Holmes MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal ’programming’ of adult pathophysiology. Nat Clin Pract Endocrinol Metab.2007 Jun; 3(6):479-88.
- Reynolds RM, Walker BR, Syddall HE,Andrew R,Wood PJ,Whorwood CB, et al. Altered control of cortisol secretion in adult men with low birth weight and cardiovascular risk factors. J Clin Endocrinol Metab 2001; 86: 245–250.
- Rich-Edwards JW, Spiegelman D, Hibert L, N E, Jun H-J, Todd, et al. Abuse in childhood and adolescence as a predictor of type 2 diabetes in adult women. American Journal of Preventive Medicine. Elsevier; 2010 Dec 1;39(6):529–36.
- 40. Boynton-Jarrett R, Rosenberg L, Palmer JR, Boggs DA, Wise LA. Child and adolescent abuse in relation to obesity in adulthood: The black women’s health study. PEDIATRICS. American Academy of Pediatrics (AAP); 2012 Jul 2;130(2):245–53.
- Johnson SB, Riley AW, Granger DA, Riis J. The science of early life toxic stress for pediatric practice and advocacy. PEDIATRICS. American Academy of Pediatrics (AAP); 2013 Jan 21;131(2):319–27.
- Madsen NJ, Kertes DA, Gunnar MR, Long JD. Early deprivation and home basal cortisol levels: A study of internationally adopted children. Development and Psychopathology. C: Cambridge University Press; 2008 Mar 1;20(02):473–91.
- Szyf M. The early life environment and the epigenome. Biochimica et Biophysica Acta (BBA) - General Subjects. Elsevier BV; 2009 Sep;1790(9):878–85.
- Shalev I. Early life stress and telomere length: Investigating the connection and possible mechanisms. BioEssays. WILEYâ€ÂÂÂÂVCH Verlag; 2012 Nov 1. 34(11):943–52.
- Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology. Nature Publishing Group; 2010 Sep 29;35(13):2617–23.
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Int J Cardiol.2014 Jun 15; 174(2):458-61.
- Smith F, Garfield A, Ward A. Regulation of growth and metabolism by imprinted genes. Cytogenetic and genome research. 2006 Apr 1;(113).
- Waterland R, Jirtle R. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Molecular and cellular biology. 2003 Jul 16 ;15(23).
- Kremer D, Lindemans J, Siebel C, Heijmans BT, Steegers-Theunissen RP, Obermann-Borst SA, et al. Periconceptional maternal Folic acid use of 400 µg per day is related to increased Methylation of the IGF2 gene in the very Young Child. PLOS ONE. Public Library of Science; 2009 Nov 16;4(11):7845.
- Jacob KJ, Robinson WP, Lefebvre L. Beckwith-Wiedemann and Silver- Russell syndromes: opposite developmental imbalances in imprinted regulators of placental function and embryonic growth. Clin Genet. 2013;84(4):326–34.
- Edwards CA, Ferguson-Smith AC.Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol. 2007;19(3):281–9.
- Schneider E, Pliushch G, ElHajj N, Galetzka D, Puhl A, Schorsch M, et al. Spatial, temporal and interindividual epigenetic variation of functionally important DNA methylation patterns. Nucleic Acids Res. 2010;38(12):3880–90.
- Cordeiro A , Neto AP, Carvalho F, Ramalho C, Dória S. Relevance of genomic imprinting in intrauterine human growth expression of CDKN1C, H19, IGF2, KCNQ1 and PHLDA2 imprinted genes. J Assist Reprod Genet.2014 Oct;31(10):1361-8. doi: 10.1007/s10815-014-0278-0. Epub 2014 Jul 2.
- Fogo A, Ichikawa L (1996) Renin and angiotensin system in development of mice and men. Am J Pathol. 149: 1797–801.
- Carey RM, Siragy HM (2003) Newly recognized components of the Renin- Angiotensin System: Potential roles in cardiovascular and renal regulation. Endocrine Rev. 24: 261–271.
- Miyawaki M, Okutani T, Higuchi R, Yoshikawa N (2006) Plasma angiotensin II concentrations in the early neonatal period. Arch Dis Child Fetal Neonatal Ed. 91: 359–362.
- Cambien F, Costerousse O, Tiret L, Poirier O, Lecerf L, Gonzales M, et al. Plasma level and gene polymorphism of angiotensin-converting enzyme in relation to myocardial infarction. Circulation. 1994 Aug 1;2(90)
- 58. Rangel M, dos Santos JC, Ortiz PH, Hirata M, Jasiulionis MG, Araujo RC,et al. Modification of epigenetic patterns in low birth weight children: importance of hypomethylation of the ACE genepromoter. PLoS One. 2014 Aug 29;9(8):e106138.
- Henry JA, Bolla M, Osmond C, Fall C, Barker DJ, Humphries SE.The effects of genotype and infant weight on adult plasma levels of fibrinogen, factor VII, and LDL cholesterol are additive.J Med Genet.1997 Jul; 34(7):553-8.
- Barker DJ, Meade TW, Fall CH, Lee A, Osmond C, Phipps K, et al. Relation of fetal and infant growth to plasma fibrinogen and factor VII concentrations in adult life. BMJ.1992 Jan 18; 304(6820):148-52.
- Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr.1999;69:179-97.
- Gallou-Kabani C, Junien C. Nutrional epigenomics of metabolic síndrome: new perspective against the epidemic. Diabetes 2005 Jul; 54 (7): 1899-906.
- Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, et al. Long- term Angiotensin II AT1 receptor inhibition produces adipose tissue hypotrophy accompanied by increased expression of adiponectin and PPARγ. Eur J Pharmacol. 2006 Dec 15;552(1-3):112-22.
- Pedersen L, Hojman P. Muscle-to-organ cross talk mediated by myokines. Adipocyte.2012 Jul 1; 1(3):164-167.
- Pedersen BK, Akerström TC, Nielsen AR, Fischer CP. Role of myokines in exercise and metabolism. J Appl Physiol, 2007. 103(3): p. 1093-8.
- Peterson MD, Saltarelli WA, Visich PS, Gordon PM. Strength capacity and cardiometabolic risk clustering in adolescents. Pediatrics. 2014 Apr; 133(4):e896- 903.
- Artero EG, Ruiz JR, Ortega FB, España-Romero V, Vicente-Rodríguez G, Molnar D, et al. Muscular and cardiorespiratory fitness are independently associated with metabolic risk in adolescents: the HELENA study. Pediatr Diabetes. 2011 Dec; 12(8):704-12.
- Bohannon RW. Muscle strength: clinical and prognostic value of hand-grip dynamometry. Curr Opin Clin Nutr Metab Care. 2015 Sep;18(5):465-70.
- Cohen DD. Neuromuscular performance deficits in low birthweight children: a target for physical activity interventions? Dev Med Child Neurol. 2015 May; 57(5):406-7.
- Malenfant P, Joanisse DR, Thériault R, Goodpaster BH, Kelley DE, Simoneau JA. Fat content in individual muscle fiber of lean and obese subjects. Int J Obes Relat Metab Disord, 2001.25 (9): p 1316-2.
- Wells JC, Chomtho S, Fewtrell MS. Programming of body composition by early growth and nutrition. Proc Nutr Soc, 2007. 66(3): p. 423-34.
- Katzmarzyk PT, Leandro, Góis C, André M, Manhãesâ€ÂÂÂÂDeâ€ÂÂÂÂCastro R, Almeida D, et al. Birthweight, body composition, and motor performance in 7â€Â to 10 year old children. Developmental Medicine & Child Neurology [Internet]. 2015 May 1 [cited 2015 Nov 19];57(5):470–5.
- Ortega FB, Labayen I, Ruiz JR, Martin-Matillas M, Vicente-Rodríguez G, Redondo C, et al. Are muscular and cardiovascular fitness partially programmed at birth? Role of body composition. The Journal of Pediatrics. Elsevier BV; 2009 Jan;154(1):61–6.e1
- Maltin CA. Muscle development and obesity: Is there a relationship? Organogenesis, 2008. 4(3): p. 158-69.
- Putman CT, Xu X, Gillies E, MacLean IM, Bell GJ. Effects of strength, endurance and combined training on myosin heavy chain content and fibre-type distribution in humans. Eur J Appl Physiol, 2004. 92(4-5): p.376-84.
- Putman CT, Xu X,Gillies E, MacLean IM, Bell GJ.Maternal nutrient restriction affects properties of skeletal muscle in offspring. Eur J Appl Physiol.2004 Aug; 92(4-5):376-84.
- Jensen CB, Storgaard H, Madsbad S, Richter EA, Vaag AA. Altered skeletal muscle fiber composition and size precede whole-body insulin resistance in young men with low birth weight. J Clin Endocrinol Metab, 2007. 92(4): p. 1530-4.
- Lillioja S, Young AA, Culter CL, Ivy JL, Abbott WG, Zawadzki JK, et al. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest, 1987. 80(2): p. 415-24.
- 79. Kim SG, Buel GR, Blenis J. Nutrient regulation of the mTOR complex 1 signaling pathway. Mol Cells 2013 Jun; 35 (6): 463-73.
- 80. Kim SG, Buel GR, Blenis J. Nutrient regulation of the mTOR complex 1 signaling pathway. Mol Cells 2013 Jun; 35 (6): 463-73.
- Daval J-L, Guéant J-L, Guéant-Rodriguez R-M, Ziegler O, Coelho D, Feigerlova E, et al. Nutritional models of foetal programming and nutrigenomic and epigenomic dysregulations of fatty acid metabolism in the liver and heart. Pflügers Archiv - European Journal of Physiology. Springer Berlin Heidelberg; 2013 Sep 3;466(5):833–50.
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007 Jun 29; 129(7):1261-74.
- Yu MX, Shen ZY, Qiu XS, Mo QP. High protein diets alters body composition and improves insulin resistance in a rat model of low weight birth. J Investig Med. 2012 Dec; 60(8):1174-9.
- Ozanne SE, Storgaard H, Jensen CB, Martin-Gronert MS, Madsbad S, Vaag A. Altered PI3-Kinase/Akt signalling in skeletal muscle of young men with low birth weight. PLOS ONE. Public Library of Science; 2008 Nov 17;3 (11):3738.
- Victora C, Sibbritt D, Horta B, Lima R, Cole T, Wells J. Weight gain in childhood and body composition at 18 years of age in Brazilian males. Acta paediatrica (Oslo, Norway : 1992). 2007 Apr 13;2 (96).

