Julie Briot1,2,4, Nazzareno D’Avanzo1,2, Jurgen Sygusch2,3 and Lucie Parent1,2,4*
1Département de Physiologie Moléculaire et Intégrative, Faculté de Médecine, Canada
2Groupe d’étude des Protéines membranaires, Canada
3Département de Biochimie et Médecine Moléculaire, Faculté de Médecine, Université de Montréal, Montréal, Québec, Canada
4Centre de Recherche de l’Institut de Cardiologie de Montréal, 5000 rue Bélanger, Montréal, Québec, Canada
Corresponding Author:
Dr. Lucie Parent
Département de Physiologie Moléculaire et Intégrative, Université de Montréal, Centre de Recherche de l’Institut de Cardiologie de Montréal, 5000 rue Bélanger, Montréal, Québec, H1T 1C8, Canada
Tel: 514-343-6673
E-mail: lucie.parent@umontreal.ca
Received date: July 01, 2016; Accepted date: September 18, 2016; Published date: September 23, 2016
Citation: Briot J, D’Avanzo N, Sygusch J, et al. Three-Dimensional Architecture of the L-Type Calcium Channel: Structural Insights into the CaVa2d1 Auxiliary Protein. Biochem Mol Biol J. 2016, 2:3. doi: 10.21767/2471-8084.100026
Keywords
L-type calcium channel; Complexes architecture; CaVa2d1 subunit; Electro-microscopy; Small angle X-ray scattering; Template-base modelling
Abbreviations
Cryo-EM: Cryo-Electron Microscopy; 3-D: Three Dimensional, PDB: Protein Data Bank; SAXS: Small Angle X-Ray Scattering; LTCC: L-Type Calcium Channel
Introduction
L-type calcium channels (LTCC) form a large family of structurally related channels expression in skeletal muscle (CaV1.1), working myocardium (CaV1.2), neuroendocrine cells (CaV1.3), and the retina (CaV1.4). In cardiac cells, calcium ions entering into the cell through CaV1.2 channels during the plateau phase of the action potential are essential to initiate the excitation contraction coupling [1,2]. Together with voltage-gated sodium and potassium channels, CaV1.2 contributes to the heart rhythm and its activity can be derived from the measure of the QT interval on the electro cardiogram [3]. Gain-of-function and lossof -function genetic mutations of sodium and potassium channels have been associated with many forms of cardiac arrhythmias [4]. To a smaller extent, mutations in the genes encoding for CaV1.2 channels have been associated with Timothy, Brugada, and early after depolarization syndromes [5-7]. Some of these cardiac dysfunctions are characterized by an increase in the QT interval, whereas others are manifested by a shorter QT interval and an elevated ST segment [4,8].
In cardiomyocytes, CaV1.2 is an oligomer consisting of a main pore-forming CaVa1 (˜ 250 kDa) and additional auxiliary subunits: CaVß2 (˜ 55 kDa) and CaVa2d1 (˜ 150 kDa). The CaVa1 subunit confers the biophysical and pharmacological properties of the channel and is the molecular target of the class IV antiarrhythmic drugs, among which dihydropyridine, verapamil, and diltiazem compounds are most widely used. This subunit is essential and targeted disruption of CaVa1 is embryonically lethal in mice [9]. The intracellular CaVß promotes cell surface trafficking of CaV1.2 through a nanomolecular interaction between the guanylate kinase domain of CaVß and the hydrophobic residues of the a-helix formed in the cytoplasmic loop of the CaVa1 subunit of CaV1.2 [10]. CaVa2d1 increases peak current density and stabilizes the channel open state [6,11]. All three subunits are required to reproduce the biophysical properties of the native channel. Over the last 15 years, structural studies have revealed the high affinity interaction between CaVß and CaVa1 as well as the Ca2+/ calmoduline-CaVa1 association by X-ray crystallography [12-15]. By contrast, there is little structural information on CaVa2d1. The reason can be found in the complexity of the CaVa2d1 protein topology that results from multiple co- and post-translational modifications including the addition of N-glycans at 16 Asn sites that is required for the folding and stability of CaVa2d1 [16,17]. Furthermore, CaVa2d1 is encoded by a single gene and is posttranslationnally cleaved into the large extracellular CaVa2 and the putative transmembrane CaVd proteins bound by disulfide bridges [18-20]. In fact, the rat CaVa2d1 protein includes 20 cysteine residues and it has been proposed that intra-molecular disulfide bonds are required to stabilize its higher order structure [21]. These features represent significant hurdles for expressing and purifying the protein complex in bacterial systems and account for the limited structural information on eukaryotic LTCC channels
Three-Dimensional Structure of the Mammalian CaVa2d1 Proteins
Structural data on the pore-forming subunit CaVa1 has been mostly inferred from the high-resolution crystal structures (˜ 2.7 – 3.1 Å) of bacterial homologs of voltage-gated sodium channels and a modified variant referred to as Ca2+-selective CaVAb channels [22-24]. Unlike their mammalian homologues, the bacterial channels form symmetrical channels with 4 identical subunits forming the pore region and appear to be functional without specific auxiliary subunits. Low-resolution electron microscopic (EM) models of the LTCC complex purified from skeletal muscle at a concentration of 90-140 µg LTCC complex for 400 g of skeletal muscle were first solved at 27 Å in 2004 [25]. The relatively low resolution provided a general outline of the protein complex and confirmed that CaVa2 was mostly extracellular. More recently improvements in the electron detection and image processing algorithms made it possible to reconstruct the three-dimensional structure of the endogenous CaV1.1 channel complex from rabbit skeletal muscle membranes [17] without the need to obtain crystals. Instead of photoaffinitylabeling the protein complex with radioactive dihydropyridine receptor ligands [26], the authors chose the brilliant strategy to overexpress the cytosolic CaVß1a subunit as a fusion protein in a bacterial system and use it as a bait to pull down with nanomolar affinity the entire LTCC complex [17,27]. Aliquots (4 µl) of the digitonin-purified Cav1.1 complex at 0.1 mg/ml were examined by cryo-EM and more than 106 particles were selected for further analysis. A three-dimensional structure of the LTCC complex with dimension of 170 x 100 Å was obtained at 4.2 Å [17] and then at 3.6 Å in the presence of 10 mM Ca2+ [27]. The electron density map revealed the position of the three subunits (CaVß1a, CaV?1, and CaVa2d1) in relation to the pore-forming CaVa1 subunit of the CaV1.1 complex. In particular, the 3-D structure demonstrates for the first time the position of the transmembrane CaV?1 protein, which is the major isoform expressed in the skeletal muscle [28]. More importantly, the authors provide the first description of the extracellular structural domains within CaVa2d1 and their position relative to CaVa1.
A BLASTP search conducted with the “conserved domain” tool in NCBI [29] revealed four structural domains in the extracellular region of the rabbit (NP_001075745.1) and the rat (NP_037051.2) CaVa2d1 proteins: i.e., VWA-N, VWA, Cache2, and VGCC, the latter being included in the Cache2 domain identified in the cryo-EM structure of the rabbit CaVa2d1 (Figure 1). The VWA domain, believed to be the molecular target of anti-epileptic drugs of the gabapentin family [30], appears to be positioned just above the voltage sensor of the CaVa1 subunit suggesting that CaVa2d1 could modulate the channel function by stabilizing the channel voltage sensor domain [31]. There was however insufficient electron density to support amino acid assignment for the region between amino acids 627 to 950 within the unstructured C-terminal domain of CaVa2 as well as for the CaVd transmembrane domain between 1065 and 1106 in the first structure at 4.2 Å. Unfortunately this relatively low resolution precluded assignment of the side chains even in regions where the backbone has been solved confidently such as in the N-terminal of CaVa2 (between 40 and 77 and between 112 and 178).
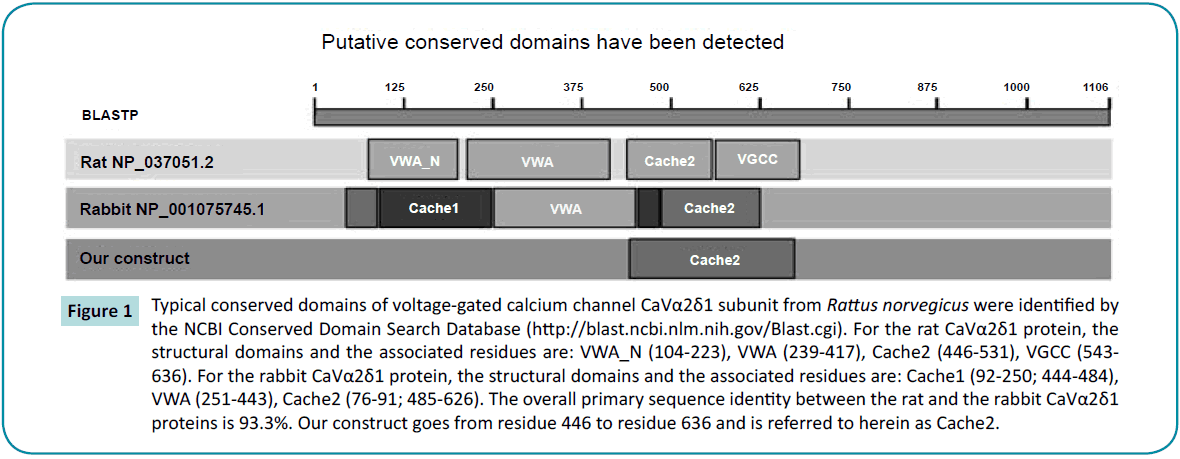
Figure 1: Typical conserved domains of voltage-gated calcium channel CaVα2δ1 subunit from Rattus norvegicus were identified by the NCBI Conserved Domain Search Database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). For the rat CaVα2δ1 protein, the structural domains and the associated residues are: VWA_N (104-223), VWA (239-417), Cache2 (446-531), VGCC (543- 636). For the rabbit CaVα2δ1 protein, the structural domains and the associated residues are: Cache1 (92-250; 444-484), VWA (251-443), Cache2 (76-91; 485-626). The overall primary sequence identity between the rat and the rabbit CaVα2δ1 proteins is 93.3%. Our construct goes from residue 446 to residue 636 and is referred to herein as Cache2.
The Cache domains face the extracellular environment and projects approximately 60 Å away from the membrane where it could anchor an extracellular networking hub for LTCC. Structural information could help identify crucial partners for protein trafficking and/or function [32]. Few mammalian homologs of the Cache2 domain are known. The primary sequence of the rat Cache2 domain of CaVa2d1 share 24% and 34% identity in their primary sequences with the sensor domain of the Bacillus subtillis histidine kinase KinD (NP_389249.1) and the Cache domain of the methyl-accepting chemotaxis protein from Methanosarcina mazei (GI:295789445) respectively according to the local alignment search tool (blast) from the PDB database [33]. This relatively low homology and the small number of templates highlight the challenges of elucidating the three-dimensional structure of the CaVa2d1 protein. The latter might prove to be a slightly superior template because the Cache domain of methylaccepting chemotaxis protein possesses the same number of residues as the Cache2 domain of the rat CaVa2d1 protein thus introducing no gap in the alignment. This contrasts with a gap of 11 residues with the former protein which accounts in part for the lower sequence identity. The online server I-TASSER [34-36] nonetheless confirmed these two proteins as the most appropriate templates for Cache2 domain of CaVa2d1 suggesting that these proteins may share similar folding patterns.
The structural complexity of CaVa2d (disulfide bonds, multiple glycosylation sites, and one transmembrane domain) makes purification of the whole protein in a bacterial system quite challenging. To bypass these limitations, we implemented the ”Divide and Conquer” approach [37] whereby the Cache2 domain, one of the basic building units of CaVa2d1 [38], was cloned into a bacterial expression vector and purified for structure determination.
SAXS Structure of the Cache2 Domain of CaVa2d1
The primary sequence of the rat Cache2 domains (between residues 446 and 636) display 93% identity with the rabbit isoform (between residues 448 and 651) (Figure 1). However, the rabbit Cache2 domain in the cryo-EM structure includes residues numbered 76 to 91 that are absent in our protein [38]. We overexpressed the Cache2 protein (rat residues 446-636) as a 25- kDa (HIS)6-tagged protein in E. coli BL21(DE3)pLYS. The protein was purified using a three-step purification procedure including affinity, DEAE and size exclusion chromatography. Most of the expressed protein found in inclusion bodies, was solubilized using 8 M urea and purified under denaturing conditions by immobilized metal ion affinity chromatography (IMAC) using a nickel resin followed by diethylaminoethanol anion-exchange chromatography. Refolding was achieved by removing urea by overnight dialysis at 4°C in the presence of 0.5 M of L-arginine, a stabilizing agent [39] and 1 mM DTT [40]. The Cache2 protein eluted as a single symmetric peak at 15.9 ml thus having an apparent molecular weight of 21 kDa according to our gel filtration calibrations. This validated the monomeric state of the purified Cache2 protein. The secondary structure of the purified domain contained 8% alpha helix and 41% beta strands. The protein was stable at 4°C at a pH of 7.4 for up to 2 weeks although N-glycosylation of eukaryotic proteins is generally not adequately performed in bacteria [41].
Structural characterization of the rat Cache2 protein was carried out using SAXS (small angle X-ray scattering) to assess the authenticity of the structure of the refolded protein. SAXS is a powerful method that provides structural information of proteins in solution [42], and requires small volumes (20-50 µl) of sample at relatively low protein concentration (0.1-1 mg/ml). SAXS provides information as to the folded/unfolded state of a protein, its aggregation, flexible domains, oligomeric state, shape and limited conformational data without any mass limitation [43]. When combined with biochemical knowledge and/or known atomic structures of component domains, SAXS provides an overall solution structure using flexible linkers to connect the structural domains [44] thus delivering a first approximation of the molecular shape, protein assembly and structural dynamics of biological macromolecules in their native state [44-46]. The program SAXSTER [47], an on-line service of the I-TASSER server, was used to build an ab initio model structure using the amino acid sequence of the rat Cache2 domain and was combined with the SAXS data as a constraint. The resulting model was then assessed against the coordinates of the Cache2 domain identified in the CaVa2d ?? structure. SAXS data were collected on the refolded rat Cache2 protein at protein concentrations of 3.2, 1.6, 0.8 and 0.4 mg /ml (Figure 2). The data collected at 3.2 mg /ml showed evidence of protein-protein interactions at very small angles and were thus excluded from further analysis. The Guinier plots (lnI(q) versus q2) were linear for the remaining concentrations at very small scattering angle (q * Rg<1.3) indicative of sample monodispersity. The ab initio molecular envelopes of the rat Cache2 protein were reconstructed and averaged in DAMMIN [48]. The resulting envelope was beanshaped and slightly asymmetrical (Fig. 2) with dimensions of 71× 51× 20 Å corresponding to the Dmax of 70 Å obtained from the pair distance probability plot [49]. The atomic coordinates of the rabbit Cache2 domain (rabbit amino acids from 448 to 651) were superimposed onto the experimental envelope using SUPCOMB program. CRYSOL was then used to calculate the solution scattering of the atomic structure of the rabbit Cache2 which was then used to fit our experimental SAXS data [50]. The discrepancy was evaluated with a chi-square value of 1.14 (Figure 3A). The analysis was performed using a Cache2 model from SAXSTER and when superimposed onto the experimental envelope, an improved chi-square value of 1.05 as illustrated by comparing panels A and B in Figure 3.
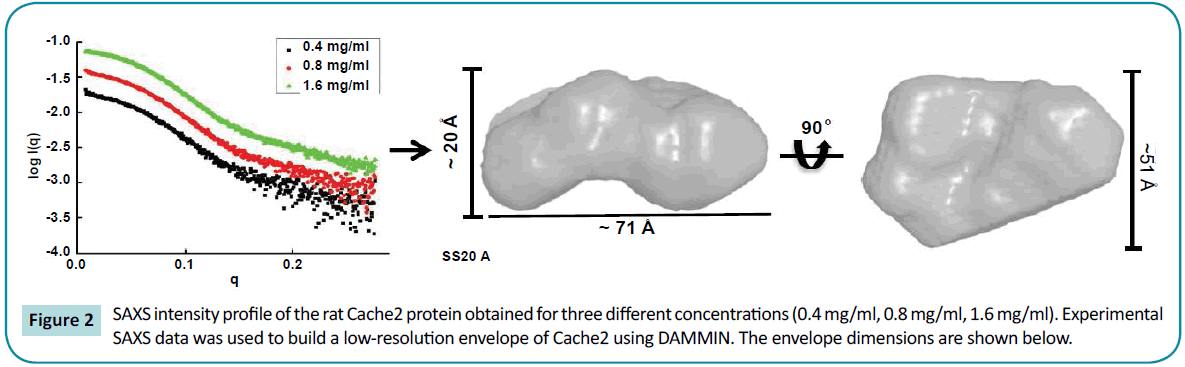
Figure 2: SAXS intensity profile of the rat Cache2 protein obtained for three different concentrations (0.4 mg/ml, 0.8 mg/ml, 1.6 mg/ml). Experimental SAXS data was used to build a low-resolution envelope of Cache2 using DAMMIN. The envelope dimensions are shown below.
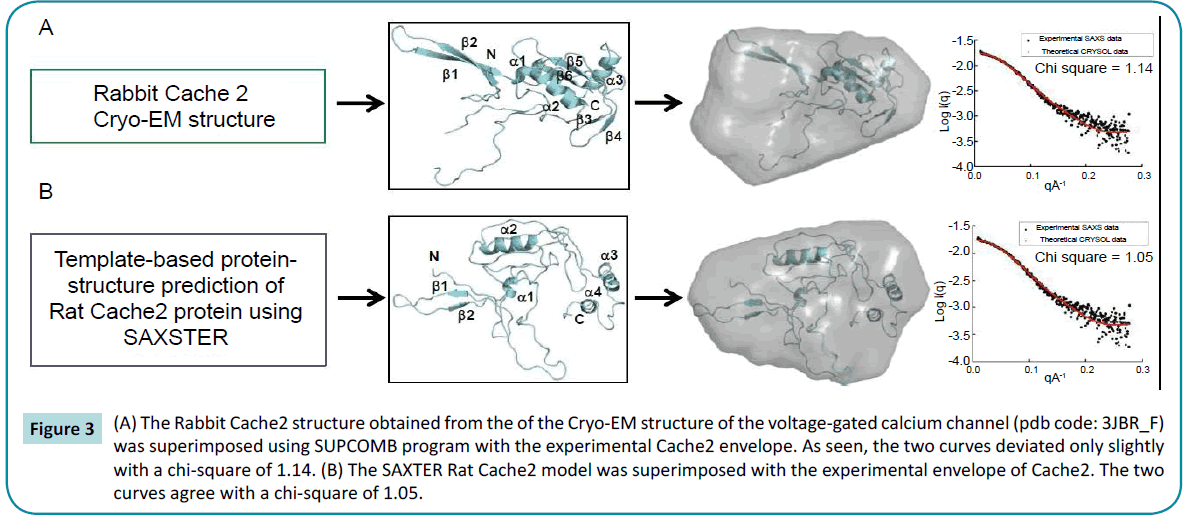
Figure 3: (A) The Rabbit Cache2 structure obtained from the of the Cryo-EM structure of the voltage-gated calcium channel (pdb code: 3JBR_F) was superimposed using SUPCOMB program with the experimental Cache2 envelope. As seen, the two curves deviated only slightly with a chi-square of 1.14. (B) The SAXTER Rat Cache2 model was superimposed with the experimental envelope of Cache2. The two curves agree with a chi-square of 1.05.
The refolded rat structure is shown to superimpose well with the rabbit EM with a Root Mean Square Deviation for C-alpha carbons = 1.73 Å (Figure 4). In particular, the structured N-terminal ß-strands 1 and 2 as well as the a-helix 1 and 2 located downstream display the best fits. The “cores” of the rabbit Cache2 and the rat Cache2 proteins are identical. Structural differences occur in the intervening loop between the amino acids 532 and 551 of the rabbit Cache2 and the rat Cache2 proteins presumably due to differences in the primary sequence between the two isoforms and due to seven additional amino acids in the primary sequence of the rat Cache2 a C-terminus a TEV cleavage site, and 6 histidine residues. The SAXS envelope from the rat Cache2 refolded protein was also well fitted by the molecular coordinates of the KinD/methyl-accepting chemotaxis protein and consistent with an ambiguous score of 2.3 obtained when performing the AMBIMETER calculation [51].
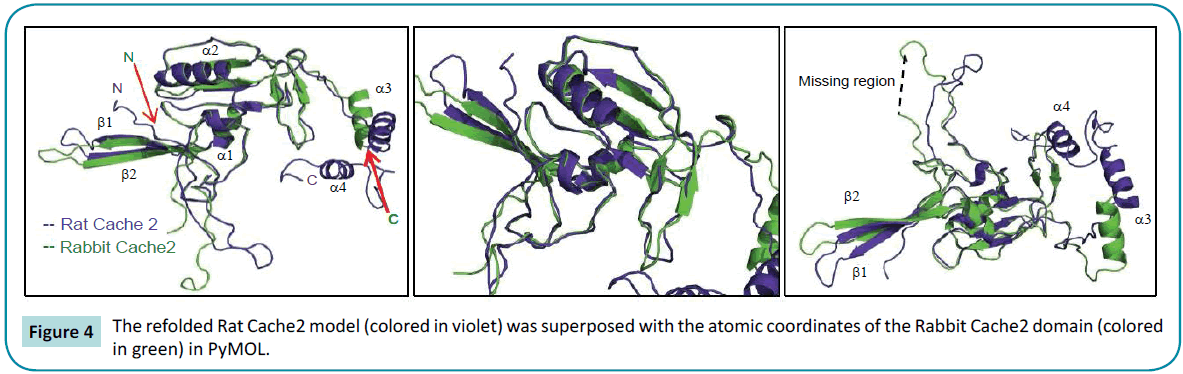
Figure 4: The refolded Rat Cache2 model (colored in violet) was superposed with the atomic coordinates of the Rabbit Cache2 domain (colored in green) in PyMOL.
Conclusion
CaVa2d1 is an integrin-like protein that belongs to the LTCC complex. It promotes LTCC activation and as a result it enhances heart contractility [11]. CaVa2d1 undergoes many co- and posttranslational modifications that create a sizable challenge for its purification. In this short commentary, we have shown that the Cache2 domain of CaVa2d1 can be purified and refolded from bacterial cultures at a yield of 2 mg per liter. SAXS data measured for the refolded protein enabled an ab initio prediction of a model structural whose fold was identical to the native state. The refolded Cache2 protein conserved the overall folding of the Cache2 protein purified from rabbit skeletal muscle, even though CaVa2d1 exists mostly as a large extracellular domain loosely organized around multiple ß-sheets [52]. Altogether, this validates an experimental strategy based upon the purification of isolated domains of LTCC subunits in a bacterial system and paves the way for implementing a “building block” approach in studying the structural biology of complex membrane proteins.
References
- Catterall WA (2011) Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3: a003947.
- Shaw RM, Colecraft HM (2013) L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc Res 98: 177-186.
- Moss AJ, Kass RS, Long QT (2005) Syndrome: from channels to cardiac arrhythmias. J Clin Invest 115: 2018-2024.
- Napolitano C, Antzelevitch C (2011) Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circulation research 108: 607-618.
- Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpon E, Hu D, et al. (2010) Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. HeartRhythm 7: 1872-1882.
- Bourdin B, Shakeri B, Tetreault MP, Sauve R, Lesage S, et al. (2015) Functional characterization of CaValpha2delta mutations associated with sudden cardiac death. J Biol Chem 290: 2854-2869.
- Raybaud A, Dodier Y, Bissonnette P, Simoes M, Bichet DG, et al. (2006) The role of the GX9GX3G motif in the gating of high voltage-activated Ca2+ channels. J Biol Chem 281: 39424-39436.
- Fukuyama M, Ohno S, Wang Q, Kimura H, Makiyama T, et al. (2013) L-type calcium channel mutations in Japanese patients with inherited arrhythmias. Circulation 77: 1799-1806.
- Rosati B, Yan Q, Lee MS, Liou SR, Ingalls B, et al. (2011) Robust L-type calcium current expression following heterozygous knockout of the Cav1.2 gene in adult mouse heart. J Physiol 589: 3275-3288.
- Bourdin B, Marger F, Wall-Lacelle S, Schneider T, Klein H, et al. (2010) Molecular determinants of the CaVbeta-induced plasma membrane targeting of the CaV1.2 channel. J Biol Chem 285: 22853-22863.
- Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, et al. (2009) Targeted disruption of the voltage-dependent calcium channel alpha2/delta -1-subunit. Am J Physiol Heart Circ Physiol 297: H117-H124.
- Van Petegem F, Clark KA, Chatelain FC, Minor Jr DL (2004) Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429: 671-675.
- Van Petegem F, Chatelain FC, Minor Jr DL (2005) Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nature structural & molecular biology 12: 1108-1115.
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, et al. (2004) Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature 429: 675-680.
- Opatowsky Y, Chen CC, Campbell KP, Hirsch JA (2004) Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42: 387-399.
- Tetreault MP, Bourdin B, Briot J, Segura E, Lesage S, et al. (2016) Identification of glycosylation sites essential for surface expression of the cavalpha2delta1 subunit and modulation of the cardiac CaV1.2 channel activity. J Biol Chem 291: 4826-4843.
- Wu J, Yan Z, Li Z, Yan C, Lu S, et al. (2015) Structure of the voltage -gated calcium channel Cav1.1 complex. Science 350: 2395.
- Dolphin AC (2013) The alpha2delta subunits of voltage-gated calcium channels. Biochim Biophys Acta 1828: 1541-1549.
- Douglas L, Davies A, Wratten J, Dolphin AC (2006) Do voltage-gated calcium channel alpha2delta subunits require proteolytic processing into alpha2 and delta to be functional? Biochem Soc Trans 34: 894-898.
- Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, et al. (2010) The alpha2delta subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci USA 107: 1654-1659.
- Klugbauer N, Marais E, Hofmann F (2003) Calcium channel alpha2delta subunits: differential expression, function, and drug binding. J Bioenerg Biomembr 35: 639-647.
- Catterall WA, Swanson TM (2015) Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol Pharmacol 88: 141-150.
- Payandeh J, Scheuer T, Zheng N, Catterall WA (2011) The crystal structure of a voltage-gated sodium channel. Nature 475: 353-358.
- Tang L, Gamal El-Din TM, Payandeh J, Martinez GQ, Heard TM, et al. (2014) Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 505: 56-61.
- Wang MC, Collins RF, Ford RC, Berrow NS, Dolphin AC, et al. (2004) The three-dimensional structure of the cardiac L-type voltage-gated calcium channel: comparison with the skeletal muscle form reveals a common architectural motif. J Biol Chem 279: 7159-7168.
- Sharp AH, Imagawa T, Leung AT, Campbell KP (1987) Identification and characterization of the dihydropyridine-binding subunit of the skeletal muscle dihydropyridine receptor. J Biol Chem 262: 12309-12315.
- Wu J, Yan Z, Li Z, Qian X, Lu S, et al. (2016) Structure of the voltage-gated calcium channel Cav1.1 at 3.6 A resolution. Nature 537: 191-196.
- Jay SD, SB Ellis, McCue AF, Williams ME, Vedvick TS, et al. (1990) Primary structure of the gamma subunit of the DHP-sensitive calcium channel from skeletal muscle. Science 248: 490-492.
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu SN, Chitsaz F, et al. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res 43: D222-D226.
- Savalli N, Pantazis A, Sigg D, Weiss JN, Neely A, et al. (2016) The alpha2delta-1 subunit remodels CaV1.2 voltage sensors and allows Ca2+ influx at physiological membrane potentials. J Gen Physiol 148: 147-159.
- Minor Jr DL, Findeisen F (2010) Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels 4: 459-474.
- Brown JP, Dissanayake VU, Briggs AR, Milic MR, Gee NS (1998) Isolation of the [3H] gabapentin-binding protein/alpha 2 delta Ca2+ channel subunit from porcine brain: development of a radioligand binding assay for alpha 2 delta subunits using [3H] leucine. Anal Biochem 255: 236-243.
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, et al. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res 43: D222-D226.
- Yang JY, Yan RX, Roy A, Xu D, Poisson J, et al. (2015) The I-TASSER Suite: protein structure and function prediction. Nature methods 12: 7-8.
- Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725-738.
- Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9: 40.
- Gaudet R (2009) Divide and conquer: high resolution structural information on TRP channel fragments. J Gen Physiol 133: 231-237.
- Song L, Espinoza-Fuenzalida IA, Etheridge S, Jones OT, Fitzgerald EM (2015) The R-domain: Identification of an N-terminal Region of the alpha2delta -1 Subunit Which is Necessary and Sufficient for its Effects on Cav2.2 Calcium Currents. Curr Mol Pharmacol 8: 169-179.
- Baynes BM, Wang DI, Trout BL (2005) Role of arginine in the stabilization of proteins against aggregation. Biochemistry 44: 4919-4925.
- Tsumoto K, Umetsu M, Kumagai I, Ejima D, Philo JS, et al. (2004) Role of arginine in protein refolding, solubilization, and purification. Biotechnol Prog 20: 1301-1308.
- Nothaft H, Szymanski CM (2010) Protein glycosylation in bacteria: sweeter than ever. Nat Rev Microbiol 8: 765-778.
- Petoukhov MV, DI Svergun (2015) Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta crystallographica Section D, Biological crystallography 71: 1051-1058.
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, et al. (2009) Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nature Methods 6: 606- 612.
- Putnam CD, Hammel M, Hura GL, Tainer JA (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys 40: 191-285.
- Boldon L, Laliberte F, Liu L (2015) Review of the fundamental theories behind small angle X-ray scattering, molecular dynamics simulations, and relevant integrated application. Nano reviews 6: 25661.
- Putnam DK, Lowe EW, Meiler J (2013) Reconstruction of SAXS Profiles from Protein Structures. Comput Struct Biotechnol J 8: e201308006.
- Dos Reis MA, Aparicio R, Zhang Y (2011) Improving Protein Template Recognition by Using Small-Angle X-Ray Scattering Profiles. Biophysical Journal 101: 2770-2781.
- Svergun DI (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophysical Journal 77: 2896-2896.
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36: 1277-1282.
- Svergun D, Barberato C, Koch MHJ (1995) CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28: 768-773.
- Petoukhov MV, Svergun DI (2015) Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta Crystallogr 71: 1051-1058.
- Upadhyay AA, Fleetwood AD, Adebali O, Finn RD, Zhulin IB (2016) Cache domains that are homologous to, but different from PAS domains comprise the largest superfamily of extracellular sensors in prokaryotes. Plos Comput Biol 12.

